Sandimmun Oral Solution
SUMMARY OF PRODUCT CHARACTERISTICS
1 NAME OF THE MEDICINAL PRODUCT
SANDIMMUN Oral Solution
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
1 ml oral solution contains 100 mg ciclosporin.
Excipient with known effect:
Ethanol: Sandimmun oral solution contains 12.6% v/v ethanol (10.0% m/v ethanol).
For the full list of excipients see section 6.1.
3 PHARMACEUTICAL FORM
Oral solution
Yellow to brownish yellow liquid, clear or with a small amount of very fine sediment.
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
Transplantation indications Solid organ transplantation
Prevention of graft rejection following solid organ transplantation.
Treatment of transplant cellular rejection in patients previously receiving other immunosuppressive agents.
Bone marrow transplantation
Prevention of graft rejection following allogeneic bone marrow and stem cell transplantation.
Prevention or treatment of graft-versus-host disease (GVHD).
Non-transplantation indications
Endogenous uveitis
Treatment of sight-threatening intermediate or posterior uveitis of non-infectious aetiology in patients in whom conventional therapy has failed or caused unacceptable side effects.
Treatment of Beh9et uveitis with repeated inflammatory attacks involving the retina in patients without neurological manifestations.
Nephrotic syndrome
Steroid-dependent and steroid-resistant nephrotic syndrome, due to primary glomerular diseases such as minimal change nephropathy, focal and segmental glomerulosclerosis, or membranous glomerulonephritis.
Sandimmun can be used to induce and maintain remissions. It can also be used to maintain steroid-induced remission, allowing withdrawal of steroids.
Rheumatoid arthritis
Treatment of severe, active rheumatoid arthritis.
Psoriasis
Treatment of severe psoriasis in patients in whom conventional therapy is inappropriate or ineffective.
Atopic dermatitis
Sandimmun is indicated in patients with severe atopic dermatitis when systemic therapy is required.
4.2 Posology and method of administration
Posology
The dose ranges given for oral administration are intended to serve as guidelines only.
The daily doses of Sandimmun should be given in two divided doses equally distributed throughout the day. It is recommended that Sandimmun be administered on a consistent schedule with regard to time of day and in relation to meals.
Sandimmun should only be prescribed by, or in close collaboration with, a physician with experience of immunosuppressive therapy and/or organ transplantation.
Transplantation
Solid organ transplantation
Treatment with Sandimmun should be initiated within 12 hours before surgery at a dose of 10 to 15 mg/kg given in 2 divided doses. This dose should be maintained as the daily dose for 1 to 2 weeks post-operatively, being gradually reduced in accordance with blood levels according to local immunosuppressive protocols until a recommended maintenance dose of about 2 to 6 mg/kg given in 2 divided doses is reached.
When Sandimmun is given with other immunosuppressants (e.g. with corticosteroids or as part of a triple or quadruple medicinal product therapy), lower doses (e.g. 3 to 6 mg/kg given in 2 divided doses for the initial treatment) may be used.
Bone marrow transplantation
The initial dose should be given on the day before transplantation. In most cases, Sandimmun concentrate for solution for infusion is preferred for this purpose. The recommended intravenous dose is 3 to 5 mg/kg/day. Infusion is continued at this dose level during the immediate post-transplant period of up to 2 weeks, before a change is made to oral maintenance therapy with Sandimmun at daily doses of about 12.5 mg/kg given in 2 divided doses.
Maintenance treatment should be continued for at least 3 months (and preferably for 6 months) before the dose is gradually decreased to zero by 1 year after transplantation.
If Sandimmun is used to initiate therapy, the recommended daily dose is 12.5 to 15 mg/kg given in 2 divided doses, starting on the day before transplantation.
Higher doses of Sandimmun, or the use of Sandimmun intravenous therapy, may be necessary in the presence of gastrointestinal disturbances which might decrease absorption.
In some patients, GVHD occurs after discontinuation of ciclosporin treatment, but usually responds favourably to re-introduction of therapy. In such cases an initial oral loading dose of 10 to 12.5 mg/kg should be given, followed by daily oral administration of the maintenance dose previously found to be satisfactory. Low doses of Sandimmun should be used to treat mild, chronic GVHD.
Non-transplantation indications
When using Sandimmun in any of the established non-transplantation indications, the following general rules should be adhered to:
Before initiation of treatment a reliable baseline level of renal function should be established by at least two measurements. The estimated glomerular filtration rate (eGFR) by the MDRD formula can be used for estimation of renal function in adults and an appropriate formula should be used to assess eGFR in paediatric patients.
Since Sandimmun can impair renal function, it is necessary to assess renal function frequently. If eGFR decreases by more than 25% below baseline at more than one measurement, the dosage of Sandimmun should be reduced by 25 to 50%. If the eGFR decrease from baseline exceeds 35%, further reduction of the dose of Sandimmun should be considered. These recommendations apply even if the patient's values still lie within the laboratory's normal range. If dose reduction is not successful in improving eGFR within one month, Sandimmun treatment should be discontinued (see section 4.4).
Regular monitoring of blood pressure is required.
The determination of bilirubin and parameters that assess hepatic function are required prior to starting therapy and close monitoring during treatment is recommended. Determinations of serum lipids, potassium, magnesium and uric acid are advisable before treatment and periodically during treatment.
Occasional monitoring of ciclosporin blood levels may be relevant in non-transplant indications, e.g. when Sandimmun is co-administered with substances that may interfere with the pharmacokinetics of ciclosporin, or in the event of unusual clinical response (e.g. lack of efficacy or increased drug intolerance such as renal dysfunction).
The normal route of administration is by mouth. If the concentrate for solution for infusion is used, careful consideration should be given to administering an adequate intravenous dose that corresponds to the oral dose. Consultation with a physician with experience of use of ciclosporin is recommended.
Except in patients with sight-threatening endogenous uveitis and in children with nephrotic syndrome, the total daily dose must never exceed 5 mg/kg.
For maintenance treatment the lowest effective and well tolerated dosage should be determined individually.
In patients in whom within a given time (for specific information see below) no adequate response is achieved or the effective dose is not compatible with the established safety guidelines, treatment with Sandimmun should be discontinued.
Endogenous uveitis
For inducing remission, initially 5 mg/kg/day orally given in 2 divided doses are recommended until remission of active uveal inflammation and improvement in visual acuity are achieved. In refractory cases, the dose can be increased to 7 mg/kg/day for a limited period.
To achieve initial remission, or to counteract inflammatory ocular attacks, systemic corticosteroid treatment with daily doses of 0.2 to 0.6 mg/kg prednisone or an equivalent may be added if Sandimmun alone does not control the situation sufficiently. After 3 months, the dose of corticosteroids may be tapered to the lowest effective dose.
For maintenance treatment, the dose should be slowly reduced to the lowest effective level. During the remission phases, this should not exceed 5 mg/kg/day.
Infectious causes of uveitis should be ruled out before immunosuppressants can be used.
Nephrotic syndrome
For inducing remission, the recommended daily dose is given in 2 divided oral doses.
If the renal function (except for proteinuria) is normal, the recommended daily dose is the following:
- adults: 5 mg/kg
- children: 6 mg/kg
In patients with impaired renal function, the initial dose should not exceed 2.5 mg/kg/day.
The combination of Sandimmun with low doses of oral corticosteroids is recommended if the effect of Sandimmun alone is not satisfactory, especially in steroid-resistant patients.
Time to improvement varies from 3 to 6 months depending on the type of glomerulopathy. If no improvement has been observed after this time to improvement period, Sandimmun therapy should be discontinued.
The doses need to be adjusted individually according to efficacy (proteinuria) and safety, but should not exceed 5 mg/kg/day in adults and 6 mg/kg/day in children.
For maintenance treatment, the dose should be slowly reduced to the lowest effective level.
Rheumatoid arthritis
For the first 6 weeks of treatment the recommended dose is 3 mg/kg/day orally given in 2 divided doses. If the effect is insufficient, the daily dose may then be increased gradually as tolerability permits, but should not exceed 5 mg/kg. To achieve full effectiveness, up to 12 weeks of Sandimmun therapy may be required.
For maintenance treatment the dose has to be titrated individually to the lowest effective level according to tolerability.
Sandimmun can be given in combination with low-dose corticosteroids and/or non-steroidal anti-inflammatory drugs (NSAIDs) (see section 4.4). Sandimmun can also be combined with low-dose weekly methotrexate in patients who have insufficient response to methotrexate alone, by using 2.5 mg/kg Sandimmun in 2 divided doses per day initially, with the option to increase the dose as tolerability permits.
Psoriasis
Sandimmun treatment should be initiated by physicians with experience in the diagnosis and treatment of psoriasis. Due to the variability of this condition, treatment must be individualised. For inducing remission, the recommended initial dose is
2.5 mg/kg/day orally given in 2 divided doses. If there is no improvement after 1 month, the daily dose may be gradually increased, but should not exceed 5 mg/kg. Treatment should be discontinued in patients in whom sufficient response of psoriatic lesions cannot be achieved within 6 weeks on 5 mg/kg/day, or in whom the effective dose is not compatible with the established safety guidelines (see section 4.4).
Initial doses of 5 mg/kg/day are justified in patients whose condition requires rapid improvement. Once satisfactory response is achieved, Sandimmun may be discontinued and subsequent relapse managed with re-introduction of Sandimmun at the previous effective dose. In some patients, continuous maintenance therapy may be necessary.
For maintenance treatment, doses have to be titrated individually to the lowest effective level, and should not exceed 5 mg/kg/day.
Atopic dermatitis
Sandimmun treatment should be initiated by physicians with experience in the diagnosis and treatment of atopic dermatitis. Due to the variability of this condition, treatment must be individualised. The recommended dose range is 2.5 to 5 mg/kg/day given in 2 divided oral doses. If a starting dose of 2.5 mg/kg/day does not achieve a satisfactory response within 2 weeks, the daily dose may be rapidly increased to a maximum of 5 mg/kg. In very severe cases, rapid and adequate control of the disease is more likely to occur with a starting dose of 5 mg/kg/day. Once satisfactory response is achieved, the dose should be reduced gradually and, if possible, Sandimmun should be discontinued. Subsequent relapse may be managed with a further course of Sandimmun.
Although an 8-week course of therapy may be sufficient to achieve clearing, up to 1 year of therapy has been shown to be effective and well tolerated, provided the monitoring guidelines are followed.
Switching between oral ciclosporin _formulations
The switch from one oral ciclosporin formulation to another should be made under physician supervision, including monitoring of blood levels of ciclosporin for transplantation patients.
Special _ populations Patients with renal impairment All indications
Ciclosporin undergoes minimal renal elimination and its pharmacokinetics are not extensively affected by renal impairment (see section 5.2). However, due to its nephrotoxic potential (see section 4.8), careful monitoring of renal function is recommended (see section 4.4).
Non-transplantation indications
With the exception of patients being treated for nephrotic syndrome, patients with impaired renal function should not receive ciclosporin (see subsection on additional precautions in nontransplantation indications in section 4.4). In nephrotic syndrome patients with impaired renal function, the initial dose should not exceed 2.5 mg/kg/day.
Patients with hepatic impairment
Ciclosporin is extensively metabolised by the liver. An approximate 2- to 3-fold increase in ciclosporin exposure may be observed in patients with hepatic impairment. Dose reduction may be necessary in patients with severe liver impairment to maintain blood levels within the recommended target range (see sections 4.4 and 5.2) and it is recommended that ciclosporin blood levels are monitored until stable levels are reached.
Paediatric population
Clinical studies have included children from 1 year of age. In several studies, paediatric patients required and tolerated higher doses of ciclosporin per kg body weight than those used in adults.
Use of Sandimmun in children for non-transplantation indications other than nephrotic syndrome cannot be recommended (see section 4.4).
Elderly population (age 65 years and above)
Experience with Sandimmun in the elderly is limited.
In rheumatoid arthritis clinical trials with ciclosporin, patients aged 65 or older were more likely to develop systolic hypertension on therapy, and more likely to show serum creatinine rises >50% above the baseline after 3 to 4 months of therapy.
Dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or medication and increased susceptibility for infections.
Method of administration Oral administration
Sandimmun oral solution should be diluted in a glass (not plastic) container with cold chocolate drink, milk, fruit juice or cola immediately before being taken, stirred well and drunk at once. Owing to its possible interference with the cytochrome P450 enzyme system, grapefruit juice should be avoided for dilution (see section 4.5). The syringe should not come in contact with the diluent. The glass must be rinsed well with some more diluent to ensure that all of the dose is taken. The syringe should not be rinsed, but wiped on the outside with a dry tissue to remove remaining drops of the solution (see section 6.6).
Precautions to be taken before handling or administering the medicinal product For instructions on dilution of the medicinal product before administration, see section 6.6.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
Combination with products containing Hypericum perforatum (St John's Wort) (see section 4.5).
Combination with medicines that are substrates for the multidrug efflux transporter P-glycoprotein or the organic anion transporter proteins (OATP) and for which elevated plasma concentrations are associated with serious and/or life-threatening events, e.g. bosentan, dabigatran etexilate and aliskiren (see section 4.5).
4.4 Special warnings and precautions for use
Medical supervision
Sandimmun should be prescribed only by physicians who are experienced in immunosuppressive therapy and can provide adequate follow-up, including regular full physical examination, measurement of blood pressure and control of laboratory safety parameters. Transplantation patients receiving this medicinal product should be managed in facilities with adequate laboratory and supportive medical resources. The physician responsible for maintenance therapy should receive complete information for the follow-up of the patient.
Lymphomas and other malignancies
Like other immunosuppressants, ciclosporin increases the risk of developing lymphomas and other malignancies, particularly those of the skin. The increased risk appears to be related to the degree and duration of immunosuppression rather than to the use of specific agents.
A treatment regimen containing multiple immunosuppressants (including ciclosporin) should therefore be used with caution as this could lead to lymphoproliferative disorders and solid organ tumours, some with reported fatalities.
In view of the potential risk of skin malignancy, patients on Sandimmun, in particular those treated for psoriasis or atopic dermatitis, should be warned to avoid excess unprotected sun exposure and should not receive concomitant ultraviolet B irradiation or PUVA photochemotherapy.
Infections
Like other immunosuppressants, ciclosporin predisposes patients to the development of a variety of bacterial, fungal, parasitic and viral infections, often with opportunistic pathogens. Activation of latent polyomavirus infections that may lead to polyomavirus associated nephropathy (PVAN), especially to BK virus nephropathy (BKVN), or to JC virus associated progressive multifocal leukoencephalopathy (PML), have been observed in patients receiving ciclosporin. These conditions are often related to a high total immunosuppressive burden and should be considered in the differential diagnosis in immunosuppressed patients with deteriorating renal function or neurological symptoms. Serious and/or fatal outcomes have been reported. Effective pre-emptive and therapeutic strategies should be employed, particularly in patients on multiple long-term immunosuppressive therapy.
Renal toxicity
A frequent and potentially serious complication, an increase in serum creatinine and urea, may occur during Sandimmun therapy. These functional changes are dose-dependent and are initially reversible, usually responding to dose reduction. During long-term treatment, some patients may develop structural changes in the kidney (e.g. interstitial fibrosis) which, in renal transplant patients, must be differentiated from changes due to chronic rejection. Frequent monitoring of renal function is therefore required according to local guidelines for the indication in question (see sections 4.2 and 4.8).
Hepatotoxicity
Sandimmun may also cause dose-dependent, reversible increases in serum bilirubin and in liver enzymes (see section 4.8). There have been solicited and spontaneous reports of hepatotoxicity and liver injury including cholestasis, jaundice, hepatitis and liver failure in patients treated with ciclosporin. Most reports included patients with significant co-morbidities, underlying conditions and other confounding factors including infectious complications and co-medications with hepatotoxic potential. In some cases, mainly in transplant patients, fatal outcomes have been reported (see section 4.8). Close monitoring of parameters that assess hepatic function is required and abnormal values may necessitate dose reduction (see sections 4.2 and 5.2).
Elderly population (age 65 years and above)
In elderly patients, renal function should be monitored with particular care.
Monitoring ciclosporin levels (see section 4.2)
When Sandimmun is used in transplant patients, routine monitoring of ciclosporin blood levels is an important safety measure. For monitoring ciclosporin levels in whole blood, a specific monoclonal antibody (measurement of parent compound) is preferred; a high-performance liquid chromatography (HPLC) method, which also measures the parent compound, can be used as well. If plasma or serum is used, a standard separation protocol (time and temperature) should be followed. For the initial monitoring of liver transplant patients, either the specific monoclonal antibody should be used, or parallel measurements using both the specific monoclonal antibody and the non-specific monoclonal antibody should be performed, to ensure a dosage that provides adequate immunosuppression.
In non-transplant patients, occasional monitoring of ciclosporin blood levels is recommended, e.g. when Sandimmun is co-administered with substances that may interfere with the pharmacokinetics of ciclosporin, or in the event of unusual clinical response (e.g. lack of efficacy or increased drug intolerance such as renal dysfunction).
It must be remembered that the ciclosporin concentration in blood, plasma, or serum is only one of many factors contributing to the clinical status of the patient. Results should therefore serve only as a guide to dosage in relationship to other clinical and laboratory parameters.
Hypertension
Regular monitoring of blood pressure is required during Sandimmun therapy. If hypertension develops, appropriate antihypertensive treatment must be instituted. Preference should be given to an antihypertensive agent that does not interfere with the pharmacokinetics of ciclosporin, e.g. isradipine (see section 4.5).
Blood lipids increased
Since Sandimmun has been reported to induce a reversible slight increase in blood lipids, it is advisable to perform lipid determinations before treatment and after the first month of therapy. In the event of increased lipids being found, restriction of dietary fat and, if appropriate, a dose reduction, should be considered.
Hyperkalaemia
Ciclosporin enhances the risk of hyperkalaemia, especially in patients with renal dysfunction. Caution is also required when ciclosporin is co-administered with potassium-sparing drugs (e.g. potassium-sparing diuretics, angiotensin converting enzyme (ACE) inhibitors, angiotensin II receptor antagonists) or potassium-containing medicinal products as well as in patients on a potassium rich diet. Control of potassium levels in these situations is advisable.
Hypomagnesaemia
Ciclosporin enhances the clearance of magnesium. This can lead to symptomatic hypomagnesaemia, especially in the peri-transplant period. Control of serum magnesium levels is therefore recommended in the peri-transplant period, particularly in the presence of neurological symptom/signs. If considered necessary, magnesium supplementation should be given.
Hyperuricaemia
Caution is required when treating patients with hyperuricaemia.
Live-attenuated vaccines
During treatment with ciclosporin, vaccination may be less effective. The use of live attenuated vaccines should be avoided (see section 4.5).
Interactions
Caution should be observed when co-administering ciclosporin with drugs that substantially increase or decrease ciclosporin plasma concentrations, through inhibition or induction of CYP3A4 and/or P-glycoprotein (see section 4.5).
Renal toxicity should be monitored when initiating ciclosporin use together with active substances that increase ciclosporin levels or with substances that exhibit nephrotoxic synergy (see section 4.5).
Concomitant use of ciclosporin and tacrolimus should be avoided (see section 4.5).
Ciclosporin is an inhibitor of CYP3A4, the multidrug efflux transporter P-glycoprotein and organic anion transporter proteins (OATP) and may increase plasma levels of co-medications that are substrates of this enzyme and/or transporter. Caution should be observed while co-administering ciclosporin with such drugs or concomitant use should be avoided (see section 4.5). Ciclosporin increases the exposure to HMG-CoA reductase inhibitors (statins). When concurrently administered with ciclosporin, the dosage of the statins should be reduced and concomitant use of certain statins should be avoided according to their label recommendations. Statin therapy needs to be temporarily withheld or discontinued in patients with signs and symptoms of myopathy or those with risk factors predisposing to severe renal injury, including renal failure, secondary to rhabdomyolysis (see section 4.5).
Following concomitant administration of ciclosporin and lercanidipine, the AUC of lercanidipine was increased three-fold and the AUC of ciclosporin was increased 21%. Therefore the simultaneous combination of ciclosporin and lercanidipine should be avoided. Administration of ciclosporin 3 hours after lercanidipine yielded no change of the lercanidipine AUC, but the ciclosporin AUC was increased by 27%. This combination should therefore be given with caution with an interval of at least 3 hours.
Special excipients: Ethanol
Sandimmun contains around 12% vol. ethanol. A 500 mg dose of Sandimmun contains 500 mg ethanol, equivalent to nearly 15 ml beer or 5 ml wine. This may be harmful in alcoholic patients and should be taken into account in pregnant or breast-feeding women, in patients presenting with liver disease or epilepsy, or if the patients is a child.
Additional precautions in non-transplantation indications Patients with impaired renal function (except nephrotic syndrome patients with a permissible degree of renal impairment), uncontrolled hypertension, uncontrolled infections, or any kind of malignancy should not receive ciclosporin.
Before initiation of treatment a reliable baseline assessment of renal function should be established by at least two measurements of eGFR. Renal function must be assessed frequently throughout therapy to allow dosage adjustment (see section 4.2).
Additional precautions in endogenous uveitis
Sandimmun should be administered with caution in patients with neurological Behcet's syndrome. The neurological status of these patients should be carefully monitored.
There is only limited experience with the use of Sandimmun in children with endogenous uveitis.
Additional precautions in nephrotic syndrome
Patients with abnormal baseline renal function should initially be treated with
2.5 mg/kg/day and must be monitored very carefully.
In some patients, it may be difficult to detect Sandimmun-induced renal dysfunction because of changes in renal function related to the nephrotic syndrome itself. This explains why, in rare cases, Sandimmun-associated structural kidney alterations have been observed without increases in serum creatinine. Renal biopsy should be considered for patients with steroid-dependent minimal-change nephropathy, in whom Sandimmun therapy has been maintained for more than 1 year.
In patients with nephrotic syndrome treated with immunosuppressants (including ciclosporin), the occurrence of malignancies (including Hodgkin's lymphoma) has occasionally been reported.
Additional precautions in rheumatoid arthritis
After 6 months of therapy, renal function needs to be assessed every 4 to 8 weeks depending on the stability of the disease, its co- medication, and concomitant diseases. More frequent checks are necessary when the Sandimmun dose is increased, or concomitant treatment with an NSAID is initiated or its dosage increased. Discontinuation of Sandimmun may also become necessary if hypertension developing during treatment cannot be controlled by appropriate therapy.
As with other long-term immunosuppressive treatments, an increased risk of lymphoproliferative disorders must be borne in mind. Special caution should be observed if Sandimmun is used in combination with methotrexate due to nephrotoxic synergy.
Additional precautions in psoriasis
Discontinuation of Sandimmun therapy is recommended if hypertension developing during treatment cannot be controlled with appropriate therapy.
Elderly patients should be treated only in the presence of disabling psoriasis, and renal function should be monitored with particular care.
There is only limited experience with the use of Sandimmun in children with psoriasis.
In psoriatic patients on ciclosporin, as in those on conventional immunosuppressive therapy, development of malignancies (in particular of the skin) has been reported. Skin lesions not typical for psoriasis, but suspected to be malignant or pre-malignant should be biopsied before Sandimmun treatment is started. Patients with malignant or pre-malignant alterations of the skin should be treated with Sandimmun only after appropriate treatment of such lesions, and if no other option for successful therapy exists.
In a few psoriatic patients treated with Sandimmun, lymphoproliferative disorders have occurred. These were responsive to prompt discontinuation.
Patients on Sandimmun should not receive concomitant ultraviolet B irradiation or PUVA photochemotherapy.
Additional precautions in atopic dermatitis
Discontinuation of Sandimmun is recommended if hypertension developing during treatment cannot be controlled with appropriate therapy.
Experience with Sandimmun in children with atopic dermatitis is limited.
Elderly patients should be treated only in the presence of disabling atopic dermatitis and renal function should be monitored with particular care.
Benign lymphadenopathy is commonly associated with flares in atopic dermatitis and invariably disappears spontaneously or with general improvement in the disease.
Lymphadenopathy observed on treatment with ciclosporin should be regularly monitored.
Lymphadenopathy which persists despite improvement in disease activity should be examined by biopsy as a precautionary measure to ensure the absence of lymphoma.
Active herpes simplex infections should be allowed to clear before treatment with Sandimmun is initiated, but are not necessarily a reason for treatment withdrawal if they occur during therapy unless infection is severe.
Skin infections with Staphylococcus aureus are not an absolute contraindication for Sandimmun therapy, but should be controlled with appropriate antibacterial agents. Oral erythromycin, which is known to have the potential to increase the blood concentration of ciclosporin (see section 4.5), should be avoided. If there is no alternative, it is recommended to closely monitor blood levels of ciclosporin, renal function, and for side effects of ciclosporin.
Patients on Sandimmun should not receive concomitant ultraviolet B irradiation or PUVA photochemotherapy.
Paediatric use in non-transplantation indications
Except for the treatment of nephrotic syndrome, there is no adequate experience available with Sandimmun. Its use in children under 16 years of age for nontransplantation indications other than nephrotic syndrome cannot be recommended.
4.5 Interaction with other medicinal products and other forms of interaction
Drug interactions
Of the many drugs reported to interact with ciclosporin, those for which the interactions are adequately substantiated and considered to have clinical implications are listed below.
Various agents are known to either increase or decrease plasma or whole blood ciclosporin levels usually by inhibition or induction of enzymes involved in the metabolism of ciclosporin, in particular CYP3A4.
Ciclosporin is also an inhibitor of CYP3A4, the multidrug efflux transporter P-glycoprotein and organic anion transporter proteins (OATP) and may increase plasma levels of co-medications that are substrates of this enzyme and/or transporters.
Medicinal products known to reduce or increase the bioavailability of ciclosporin: In transplant patients frequent measurement of ciclosporin levels and, if necessary, ciclosporin dosage adjustment is required, particularly during the introduction or withdrawal of the co-administered medication. In non-transplant patients the relationship between blood level and clinical effects is less well established. If medicinal products known to increase ciclosporin levels are given concomitantly, frequent assessment of renal function and careful monitoring for ciclosporin-related side effects may be more appropriate than blood level measurement.
Drugs that decrease ciclosporin levels
All inducers of CYP3A4 and/or P-glycoprotein are expected to decrease ciclosporin levels. Examples of drugs that decrease ciclosporin levels are:
Barbiturates, carbamazepine, oxcarbazepine, phenytoin; nafcillin, intravenous sulfadimidine, probucol, orlistat, hypericum perforatum (St. John’s wort), ticlopidine, sulfinpyrazone, terbinafine, bosentan.
Products containing Hypericum perforatum (St John s Wort) must not be used concomitantly with Sandimmun due to the risk of decreased blood levels of ciclosporin and thereby reduced effect (see section 4.3).
Rifampicin induces ciclosporin intestinal and liver metabolism. Ciclosporin doses may need to be increased 3- to 5-fold during co-administration.
Octreotide decreases oral absorption of ciclosporin and a 50% increase in the ciclosporin dose or a switch to intravenous administration could be necessary.
Drugs that increase ciclosporin levels
All inhibitors of CYP3A4 and/or P-glycoprotein may lead to increased levels of cyclosporine. Examples are:
Nicardipine, metoclopramide, oral contraceptives, methylprednisolone (high dose), allopurinol, cholic acid and derivatives, protease inhibitors, imatinib, colchicine, nefazodone.
Macrolide antibiotics: Erythromycin can increase ciclosporin exposure 4- to 7-fold, sometimes resulting in nephrotoxicity. Clarithromycin has been reported to double the exposure of ciclosporin. Azitromycin increases ciclosporin levels by around 20%.
Azole antibiotics: Ketoconazole, fluconazole, itraconazole and voriconazole could more than double ciclosporin exposure.
Verapamil increases ciclosporin blood concentrations 2- to 3-fold.
Co-administration with telaprevir resulted in approximately 4.64-fold increase in ciclosporin dose normalised exposure (AUC).
Amiodarone substantially increases the plasma ciclosporin concentration concurrently with an increase in serum creatinine. This interaction can occur for a long time after withdrawal of amiodarone, due to its very long half-life (about 50 days).
Danazol has been reported to increase ciclosporin blood concentrations by approximately 50%.
Diltiazem (at doses of 90 mg/day) can increase ciclosporin plasma concentrations by up to 50%.
Imatinib could increase ciclosporin exposure and Cmax by around 20%.
Food interactions
The concomitant intake of grapefruit and grapefruit juice has been reported to increase the bioavailability of ciclosporin.
Combinations with increased risk_ for nephrotoxicity
Care should be taken when using ciclosporin together with other active substances that exhibit nephrotoxic synergy such as: aminoglycosides (includinggentamycin, tobramycin), amphotericin B, ciprofloxacin, vancomycin, trimethoprim (+ sulfamethoxazole); fibric acid derivatives (e.g. bezafibrate, fenofibrate); NSAIDs (including diclofenac, naproxen, sulindac); melphalan histamine H2-receptor antagonists (e.g. cimetidine, ranitidine); methotrexate (see section 4.4).
During the concomitant use of a drug that may exhibit nephrotoxic synergy, close monitoring of renal function should be performed. If a significant impairment of renal function occurs, the dosage of the co-administered medicinal product should be reduced or alternative treatment considered.
Concomitant use of ciclosporin and tacrolimus should be avoided due to the risk for nephrotoxicity and pharmacokinetic interaction via CYP3A4 and/or P-gp (see section 4.4).
Effects of ciclosporin on other drugs
Ciclosporin is an inhibitor of CYP3A4, the multidrug efflux transporter P-glycoprotein (P-gp) and organic anion transporter proteins (OATP). Co-administration of drugs that are substrates of CYP3A4, P-gp and OATP with ciclosporin may increase plasma levels of co-medications that are substrates of this enzyme and/or transporter.
Some examples are listed below:
Ciclosporin may reduce the clearance of digoxin, colchicine, HMG-CoA reductase inhibitors (statins) and etoposide. If any of these drugs are used concurrently with ciclosporin, close clinical observation is required in order to enable early detection of toxic manifestations of the medicinal products, followed by reduction of its dosage or its withdrawal. When concurrently administered with ciclosporin, the dosage of the statins should be reduced and concomitant use of certain statins should be avoided according to their label recommendations. Exposure changes of commonly used statins with ciclosporin are summarised in Table 1. Statin therapy needs to be temporarily withheld or discontinued in patients with signs and symptoms of myopathy or those with risk factors predisposing to severe renal injury, including renal failure, secondary to rhabdomyolysis.
Table 1 Summary of exposure changes of commonly used statins with ciclosporin
|
Statin |
Doses available |
Fold change in exposure with ciclosporin |
|
Atorvastatin |
10-80 mg |
8-10 |
|
Simvastatin |
10-80 mg |
6-8 |
|
Fluvastatin |
20-80 mg |
2-4 |
|
Lovastatin |
20-40 mg |
5-8 |
|
Pravastatin |
20-80 mg |
5-10 |
|
Rosuvastatin |
5-40 mg |
5-10 |
|
Pitavastatin |
1-4 mg |
4-6 |
Caution is recommended when co-administering ciclosporin with lercanidipine (see section 4.4).
Following concomitant administration of ciclosporin and aliskiren, a P-gp substrate, the Cmax of aliskiren was increased approximately 2.5-fold and the AUC approximately 5-fold. However, the pharmacokinetic profile of ciclosporin was not significantly altered. Coadministration of ciclosporin and aliskiren is not recommended (see section 4.3).
Concomitant administration of dabigatran extexilate is not recommended due to the P-gp inhibitory activity of ciclosporin (see section 4.3).
The concurrent administration of nifedipine with ciclosporin may result in an increased rate of gingival hyperplasia compared with that observed when ciclosporin is given alone.
The concomitant use of diclofenac and ciclosporin has been found to result in a significant increase in the bioavailability of diclofenac, with the possible consequence of reversible renal function impairment. The increase in the bioavailability of diclofenac is most probably caused by a reduction of its high first-pass effect. If NSAIDs with a low first-pass effect (e.g. acetylsalicylic acid) are given together with ciclosporin, no increase in their bioavailability is to be expected.
Elevations in serum creatinine were observed in the studies using everolimus or sirolimus in combination with full-dose ciclosporin for microemulsion. This effect is often reversible with ciclosporin dose reduction. Everolimus and sirolimus had only a minor influence on ciclosporin pharmacokinetics. Co-administration of ciclosporin significantly increases blood levels of everolimus and sirolimus.
Caution is required with concomitant use of potassium-sparing medicinal products (e.g. potassium-sparing diuretics, ACE inhibitors, angiotensin II receptor antagonists) or potassium-containing medicinal products since they may lead to significant increases in serum potassium (see section 4.4).
Ciclosporin may increase the plasma concentrations of repaglinide and thereby increase the risk of hypoglycaemia.
Co-administration of bosentan and ciclosporin in healthy volunteers increases the bosentan exposure several-fold and there was a 35% decrease in ciclosporin exposure. Coadministration of ciclosporin with bosentan is not recommended (see above subsection “Drugs that decrease ciclosporin levels” and section 4.3).
Multiple dose administration of ambrisentan and ciclosporin in healthy volunteers resulted in an approximately 2-fold increase in ambrisentan exposure, while the ciclosporin exposure was marginally increased (approximately 10%).
A significantly increased exposure to anthracycline antibiotics (e.g. doxorubicine, mitoxanthrone, daunorubicine) was observed in oncology patients with the intravenous coadministration of anthracycline antibiotics and very high doses of ciclosporin.
During treatment with ciclosporin, vaccination may be less effective and the use of live attenuated vaccines should be avoided.
Paediatric population
Interaction studies have only been performed in adults.
4.6 Fertility, pregnancy and lactation
Pregnancy
Animal studies have shown reproductive toxicity in rats and rabbits.
Experience with Sandimmun in pregnant women is limited. Pregnant women receiving immunosuppressive therapies after transplantation, including ciclosporin and ciclosporin-containing regimens, are at risk of premature delivery (<37 weeks).
A limited number of observations in children exposed to ciclosporin in utero are available, up to an age of approximately 7 years. Renal function and blood pressure in these children were normal. However, there are no adequate and well-controlled studies in pregnant women and therefore Sandimmun should not be used during pregnancy unless the potential benefit to the mother justifies the potential risk to the foetus. The ethanol content of the Sandimmun formulations should also be taken into account in pregnant women (see section 4.4).
Breast-feeding
Ciclosporin passes into breast milk. The ethanol content of the Sandimmun formulations should also be taken into account in women who are breast-feeding (see section 4.4). Mothers receiving treatment with Sandimmun should not breast-feed because of the potential of Sandimmun to cause serious adverse drug reactions in breast-fed newborns/infants. A decision should be made whether to abstain from breast-feeding or to abstain from using the medicinal drug, taking into account the importance of the medicinal product to the mother.
Fertility
There is limited data on the effect of Sandimmun on human fertility (see section 5.3).
4.7 Effects on ability to drive and use machines
No data exist on the effects of Sandimmun on the ability to drive and use machines.
4.8 Undesirable effects
Summary of the safety profile
The principal adverse reactions observed in clinical trials and associated with the administration of ciclosporin include renal dysfunction, tremor, hirsutism, hypertension, diarrhoea, anorexia, nausea and vomiting.
Many side effects associated with ciclosporin therapy are dose-dependent and responsive to dose reduction. In the various indications the overall spectrum of side effects is essentially the same; there are, however, differences in incidence and severity. As a consequence of the higher initial doses and longer maintenance therapy required after transplantation, side effects are more frequent and usually more severe in transplant patients than in patients treated for other indications.
Anaphylactoid reactions have been observed following intravenous administration (see section 4.4).
Infections and infestations
Patients receiving immunosuppressive therapies, including ciclosporin and ciclosporin-containing regimens, are at increased risk of infections (viral, bacterial, fungal, parasitic) (see section 4.4). Both generalised and localised infections can occur. Pre-existing infections may also be aggravated and reactivation of polyomavirus infections may lead to polyomavirus-associated nephropathy (PVAN) or to JC virus associated progressive multifocal leukopathy (PML). Serious and/or fatal outcomes have been reported.
Neoplasms benign, malignant and unspecified (including cysts and polyps)
Patients receiving immunosuppressive therapies, including ciclosporin and ciclosporin containing regimens, are at increased risk of developing lymphomas or lymphoproliferative disorders and other malignancies, particularly of the skin. The frequency of malignancies increases with the intensity and duration of therapy (see section 4.4). Some malignancies may be fatal.
Tabulated summary of adverse drug reactions from clinical trials
Adverse drug reactions from clinical trials (Table 1) are listed by MedDRA system organ class. Within each system organ class, the adverse drug reactions are ranked by frequency, with the most frequent reactions first. Within each frequency grouping, adverse drug reactions are presented in order of decreasing seriousness. In addition the corresponding frequency category for each adverse drug reaction is based on the following convention (CIOMS III): very common (>1/10); common (>1/100, <1/10); uncommon (>1/1,000, <1/100); rare (>1/10,000, <1/1,000) very rare (<1/10,000), not known (cannot be estimated from the available data).
Table 1: Adverse drug reactions from clinical trials Blood and lymphatic system disorders Common Leucopenia
Uncommon Thrombocytopenia, anaemia
Rare Haemolytic uraemic syndrome, microangiopathic haemolytic anaemia
Not known* Thrombotic microangiopathy, thrombotic thrombocytopenic purpura
Metabolism and nutrition disorders Very common Hyperlipidaemia
Common Hyperglycaemia, anorexia, hyperuricaemia, hyperkalaemia,
hypomagnesaemia Nervous system disorders Very common Tremor, headache
Common Convulsions, paraesthesia
Uncommon Encephalopathy including Posterior Reversible Encephalopathy
Rare
Very rare
Not known*
Vascular disorders Very common Common Gastrointestinal disorders
Syndrome (PRES), signs and symptoms such as convulsions, confusion, disorientation, decreased responsiveness, agitation, insomnia, visual disturbances, cortical blindness, coma, paresis and cerebellar ataxia Motor polyneuropathy
Optic disc oedema, including papilloedema, with possible visual impairment secondary to benign intracranial hypertension Migraine
Hypertension
Flushing
Common
Rare
Nausea, vomiting, abdominal discomfort/pain, diarrhoea, gingival
hyperplasia, peptic ulcer
Pancreatitis
Hepatobiliary disorders
Common Hepatic function abnormal (see section 4.4)
Not known* Hepatotoxicity and liver injury including cholestasis, jaundice,
hepatitis and liver failure with some fatal outcome (see section 4.4)
Skin and subcutaneous tissue disorders Very common Hirsutism
Common Acne, hypertrichosis
Uncommon Allergic rashes
Musculoskeletal and connective tissue disorders Common Myalgia, muscle cramps
Rare Muscle weakness, myopathy
Not known* Pain of lower extremities
Renal and urinary disorders
Very common Renal dysfunction (see section 4.4)
Reproductive system and breast disorders Rare Menstrual disturbances, gynaecomastia
General disorders and administration site conditions Common Pyrexia, fatigue
Uncommon Oedema, weight increase
* Adverse events reported from post marketing experience where the ADR frequency is not known due to the lack of a real denominator.
Other adverse drug reactions from post-marketing experience
There have been solicited and spontaneous reports of hepatotoxicity and liver injury including cholestasis, jaundice hepatitis and liver failure in patients treated with ciclosporin. Most reports included patients with significant co-morbidities, underlying conditions and other confounding factors including infectious complications and co-medications with hepatotoxic potential. In some cases, mainly in transplant patients, fatal outcomes have been reported (see section 4.4).
Acute and chronic nephrotoxicity
Patients receiving calcineurin inhibitor (CNI) therapies, including ciclosporin and ciclosporin-containing regimens, are at increased risk of acute or chronic nephrotoxicity. There have been reports from clinical trials and from the post-marketing setting associated with the use of Sandimmun. Cases of acute nephrotoxicity reported disorders of ion homeostasis, such as hyperkalaemia, hypomagnesaemia, and hyperuricaemia. Cases reporting chronic morphological changes included arteriolar hyalinosis, tubular atrophy and interstitial fibrosis (see section 4.4).
Pain of lower extremities
Isolated cases of pain of lower extremities have been reported in association with ciclosporin. Pain of lower extremities has also been noted as part of Calcineurin-Inhibitor Induced Pain Syndrome (CIPS).
Paediatric population
Clinical studies have included children from 1 year of age using standard ciclosporin dosage with a comparable safety profile to adults.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product.
Healthcare professionals are asked to report any suspected adverse reactions via Yellow Card Scheme (www.mhra.gov.uk/yellowcard).
4.9 Overdose
The oral LD50 of ciclosporin is 2,329 mg/kg in mice, 1,480 mg/kg in rats and >
1,000 mg/kg in rabbits. The intravenous LD50 is 148 mg/kg in mice, 104 mg/kg in rats, and 46 mg/kg in rabbits.
Symptoms
Experience with acute overdosage of ciclosporin is limited. Oral doses of ciclosporin of up to 10 g (about 150 mg/kg) have been tolerated with relatively minor clinical consequences, such as vomiting, drowsiness, headache, tachycardia and in a few patients moderately severe, reversible impairment of renal function. However, serious symptoms of intoxication have been reported following accidental parenteral overdosage with ciclosporin in premature neonates.
Treatment
In all cases of overdosage, general supportive measures should be followed and symptomatic treatment applied. Forced emesis and gastric lavage may be of value within the first few hours after oral intake. Ciclosporin is not dialysable to any great extent, nor is it well cleared by charcoal haemoperfusion.
5 PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Immunosuppressive agents, calcineurin inhibitors, ATC code: L04AD01
Ciclosporin (also known as ciclosporin A) is a cyclic polypeptide consisting of 11 amino acids. It is a potent immunosuppressive agent, which in animals prolongs survival of allogeneic transplants of skin, heart, kidney, pancreas, bone marrow, small intestine or lung. Studies suggest that ciclosporin inhibits the development of cell-mediated reactions, including allograft immunity, delayed cutaneous hypersensitivity, experimental allergic encephalomyelitis, Freund's adjuvant arthritis, graft-versus-host disease (GVHD), and also T-cell dependent antibody production. At the cellular level it inhibits production and release of lymphokines including interleukin 2 (T-cell growth factor, TCGF). Ciclosporin appears to block the resting lymphocytes in the G0 or G1 phase of the cell cycle, and inhibits the antigen-triggered release of lymphokines by activated T-cells.
All available evidence suggests that ciclosporin acts specifically and reversibly on lymphocytes. Unlike cytostatic agents, it does not depress haemopoiesis and has no effect on the function of phagocytic cells.
Successful solid organ and bone marrow transplantations have been performed in man using ciclosporin to prevent and treat rejection and GVHD. Ciclosporin has been used successfully both in hepatitis C virus (HCV) positive and HCV negative liver transplants recipients. Beneficial effects of ciclosporin therapy have also been shown in a variety of conditions that are known, or may be considered to be of autoimmune origin.
Paediatric population: Ciclosporin has been shown to be efficacious in steroid-dependent nephrotic syndrome.
5.2 Pharmacokinetic properties
Absorption
Following oral administration of Sandimmun peak blood concentrations are reached within 1 to 6 hours. The absolute oral bioavailability following administration of Sandimmun is 20 to 50%. The absorption of ciclosporin is variable and may be influenced by intake of food.
About 37% increase in AUC Cmax was observed when Sandimmun was administered with high-fat meal. Within the therapeutic dose range the peak plasma concentration and the area under the plasma concentration/time curve are proportional to the dose; for whole blood, however, the relationship is non-linear. Sandimmun oral solution and soft gelatin capsules are bioequivalent. The inter- and intra-subject variability ranges between 18 to 74%.
Distribution
Ciclosporin is distributed largely outside the blood volume, with an average apparent distribution volume of 3.5 l/kg. In the blood, 33 to 47% is present in plasma, 4 to 9% in lymphocytes, 5 to 12% in granulocytes, and 41 to 58% in erythrocytes. In plasma, approximately 90% is bound to proteins, mostly lipoproteins.
Biotransformation
Ciclosporin is extensively metabolised to approximately 15 metabolites. Metabolism mainly takes place in the liver via cytochrome P450 3A4 (CYP3A4), and the main pathways of metabolism consist of mono- and dihydroxylation and N-demethylation at various positions of the molecule. All metabolites identified so far contain the intact peptide structure of the parent compound; some possess weak immunosuppressive activity (up to one-tenth that of the unchanged drug).
Elimination
There is a high variability in the data reported on the terminal elimination half-life of ciclosporin, depending on the assay applied and the target population. The terminal half-life ranged from 6.3 hours in healthy volunteers to 20.4 hours in patients with severe liver disease. The excretion is primarily biliary, with only 6% of an oral dose excreted in the urine, and with less than 1% in the unchanged form (see sections 4.2 and 4.4). The elimination half-life in kidney-transplanted patients was approximately 11 hours, with a range between 4 and 25 hours.
Special populations
Patients with renal impairment
In a study performed in patients with terminal renal failure, the systemic clearance was approximately two thirds of the mean systemic clearance in patients with normally functioning kidneys. Less than 1% of the administered dose is removed by dialysis.
Patients with hepatic impairment
An approximate 2- to 3-fold increase in ciclosporin exposure may be observed in patients with hepatic impairment. In a study performed in severe liver disease patients with biopsy-proven cirrhosis, the terminal half-life was 20.4 hours (range between 10.8 to 48.0 hours) compared to 7.4 to 11.0 hours in healthy subjects.
Paediatric population
Pharmacokinetic data from paediatric patients given Sandimmun Neoral or Sandimmunare very limited. In 15 renal transplant patients aged 3 -16 years, ciclosporin whole blood clearance after intravenous administration of Sandimmun was 10.6±3.7 ml/min/kg (assay: Cyclo-trac specific RIA). In a study of 7 renal transplant patients aged 2-16 years, the ciclosporin clearance ranged from 9.8 to15.5 ml/min/kg. In 9 liver transplant patients aged 0.65-6 years, clearance was 9.3±5.4 ml/min/kg (assay: HPLC). In comparison to adult transplant populations, the differences in bioavailability between Sandimmun Neoral and Sandimmun in paediatrics are comparable to those observed in adults.
5.3 Preclinical safety data
Ciclosporin gave no evidence of mutagenic or teratogenic effects in the standard test systems with oral application (rats up to 17 mg/kg/day and rabbits up to 30 mg/kg/day orally). At toxic doses (rats at 30 mg/kg/day and rabbits at 100 mg/kg/day orally), ciclosporin was embryo- and foetotoxic as indicated by increased prenatal and postnatal mortality, and reduced foetal weight together with related skeletal retardations.
In two published research studies, rabbits exposed to ciclosporin in utero (10 mg/kg/day subcutaneously) demonstrated reduced numbers of nephrons, renal hypertrophy, systemic hypertension, and progressive renal insufficiency up to 35 weeks of age. Pregnant rats which received 12 mg/kg/day of ciclosporin intravenously (twice the recommended human intravenous dose) had foetuses with an increased incidence of ventricular septal defect. These findings have not been demonstrated in other species and their relevance for humans is unknown. No impairment in fertility was demonstrated in studies in male and female rats.
Ciclosporin was tested in a number of in vitro and in vivo tests for genotoxicity with no evidence for a clincally relevant mutagenic potential.
Carcinogenicity studies were carried out in male and female rats and mice. In the 78-week mouse study, at doses of 1, 4, and 16 mg/kg/day, evidence of a statistically significant trend was found for lymphocytic lymphomas in females, and the incidence of hepatocellular carcinomas in mid-dose males significantly exceeded the control value. In the 24-month rat study conducted at 0.5, 2, and 8 mg/kg/day, pancreatic islet cell adenomas significantly exceeded the control rate at the low dose level. The hepatocellular carcinomas and pancreatic islet cell adenomas were not dose related.
6.1 List of excipients
Ethanol anhydrous Maize oil interesterified Maize oil refined
6.2 Incompatibilities
Not applicable.
6.3 Shelf life
3 years
Use immediately after dilution
6.4 Special precautions for storage
The oral solution should not be refrigerated. It may be stored at room temperature not exceeding 30°C. A slight precipitate that may occur during storage does not affect the efficacy and safety of the drug. Once the bottle has been opened, the content must be used within 2 months.
6.5 Nature and contents of container
SANDIMMUN Oral Solution is available in 20 mL and 50 mL amber glass bottles with an aluminium cap and rubber stopper. A dispenser set is also provided. The tear-off cap indicates if the bottle has been previously opened. A white polypropylene cap is provided for closure of the bottle during the in-use period.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal
Sandimmun oral solution is provided with two syringes for measuring the doses. The 1-ml syringe is used to measure doses less than or equal to 1 ml (each graduation of 0.05 ml
corresponds to 5 mg of ciclosporin). The 4-ml syringe is used to measure doses greater than 1 ml and up to 4 ml (each graduation of 0.1 ml corresponds to 10 mg of ciclosporin).

Initial use of Sandimmun oral solution_
1. Raise the flap in the centre of the metal sealing ring.
2. Tear off the sealing ring completely.
3. Remove the black stopper and throw it away.
4. Push the tube unit with the white stopper firmly into the neck of the bottle.
5. Choose the syringe depending on the prescribed volume. For volume less than 1 ml or equal to 1 ml, use the 1-ml syringe. For volume greater than 1 ml, use the 4-ml syringe. Insert the nozzle of the syringe into the white stopper.
6. Draw up the prescribed volume of solution (position the lower part of the plunger ring in front of the graduation corresponding to the prescribed volume).
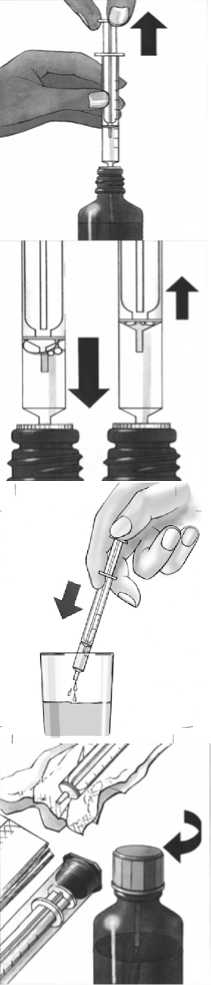
7. Expel any large bubbles by depressing and withdrawing the plunger a few times before removing the syringe containing the prescribed dose from bottle. The presence of a few tiny bubbles is of no importance and will not affect the dose in any way.
8. Push the medicine out of the syringe into a small glass with some liquid (not grapefruit juice). Avoid any contact between the syringe and the liquid in the glass. The medicine can be mixed just before it is taken. Stir and drink the entire mixture right away. Once mixed it should be taken immediately after preparation.
9. After use, wipe the syringe on the outside only with a dry tissue and replace it in its cover. The white stopper and tube should remain in the bottle. Close the bottle with the cap provided.
Subsequent use
Commence at point 5.
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
7 MARKETING AUTHORISATION HOLDER
Novartis Pharmaceuticals UK Limited Trading as SANDOZ PHARMACEUTICALS
Frimley Business Park
Frimley
Camberley
Surrey
GU16 5SG
MARKETING AUTHORISATION NUMBER(S)
PL 00101/0124
9
10
DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
17/02/1983 / 06/12/2004
DATE OF REVISION OF THE TEXT
10/06/2015