Acondro 3 Mg/0.03 Mg Film-Coated Tablets
SUMMARY OF PRODUCT CHARACTERISTICS
1 NAME OF THE MEDICINAL PRODUCT
Acondro 3 mg/0.03 mg film-coated tablets
2. QUALITATIVE AND QUANTITATIVE COMPOSITION
Each film-coated tablet contains 0.03 mg of ethinylestradiol and 3 mg of drospirenone
Excipient with known effect: Each film-coated tablet contains 62 mg lactose monohydrate.
For the full list of excipients, see section 6.1.
3 PHARMACEUTICAL FORM
Film-coated tablet.
Yellow round film-coated tablets of 5.7 mm diameter.
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
Oral contraception
The decision to prescribe Acondro should take into consideration the individual woman’s current risk factors, particularly those for venous thromboembolism (VTE), and how the risk of VTE with Acondro compares with other combined hormonal contraceptives (CHCs) (see sections 4.3 and 4.4).
4.2 Posology and method of administration
Posology
How to take Acondro
The tablets must be taken every day at about the same time, if necessary with a little liquid, in the order shown on the blister pack. One tablet is to be taken daily for 21 consecutive days. Each subsequent pack is started after a 7-day tablet-free interval, during which time a withdrawal bleed usually occurs. This usually starts on day 2-3 after the last tablet and may not have finished before the next pack is started.
How to start Acondro
• No preceding hormonal contraceptive use (in the past month)
Tablet-taking has to start on day 1 of the woman’s natural cycle (i.e. the first day of her menstrual bleeding).
• Changing from a combined hormonal contraceptive (combined oral
contraceptive (COC), vaginal ring, or transdermal patch)
The woman should start with Acondro preferably on the day after the last active tablet (the last tablet containing the active substances) of her previous COC, but at the latest on the day following the usual tablet-free or placebo tablet interval of her previous COC. In case a vaginal ring or transdermal patch has been used, the woman should start using Acondro preferably on the day of removal, but at the latest when the next application would have been due.
• Changing from a progestogen-only-method (progestogen-only pill, injection, implant)
or from a progestogen-releasing intrauterine system (IUS)
The woman may switch any day from the progestogen-only pill (from an implant or the IUS on the day of its removal, from an injectable when the next injection would be due) but should in all of these cases be advised to additionally use a barrier method for the first 7 days of tablet-taking.
• Following first-trimester abortion
The woman may start immediately. When doing so, she need not take additional contraceptive measures.
• Following delivery or second-trimester abortion
Women should be advised to start at day 21 to 28 after delivery or second-trimester abortion. When starting later, the woman should be advised to additionally use a barrier method for the first 7 days. However, if intercourse has already occurred, pregnancy should be excluded before the actual start of COC use or the woman has to wait for her first menstrual period.
For breast-feeding women see section 4.6.
Management of missed tablets
If the user is less than 12 hours late in taking any tablet, contraceptive protection is not reduced. The woman should take the tablet as soon as she remembers and should take further tablets at the usual time.
If she is more than 12 hours late in taking any tablet, contraceptive protection may be reduced. The management of missed tablets can be guided by the following two basic rules:
1. tablet-taking must never be discontinued for longer than 7 days
2. 7 days of uninterrupted tablet-taking are required to attain adequate suppression of the hypothalamic-pituitary-ovarian-axis.
Accordingly the following advice can be given in daily practice:
• Week 1
The user should take the last missed tablet as soon as she remembers, even if this means taking two tablets at the same time. She then continues to take tablets at her usual time. In addition, a barrier method such as a condom should be used for the next 7 days. If intercourse took place in the preceding 7 days, the possibility of a pregnancy should be considered. The more tablets are missed and the closer they are to the regular tablet-free interval, the higher the risk of a pregnancy.
• Week 2
The user should take the last missed tablet as soon as she remembers, even if this means taking two tablets at the same time. She then continues to take tablets at her usual time. Provided that the woman has taken her tablets correctly in the 7 days preceding the first missed tablet, there is no need to use extra contraceptive precautions. However, if she has missed more than 1 tablet, the woman should be advised to use extra precautions for 7 days.
• Week 3
The risk of reduced reliability is imminent because of the forthcoming 7-day tablet-free interval.
However, by adjusting the tablet-intake schedule, reduced contraceptive protection can still be prevented. By adhering to either of the following two options, there is therefore no need to use extra contraceptive precautions, provided that in the 7 days preceding the first missed tablet the woman has taken all tablets correctly. If this is not the case, she should follow the first of these two options and use extra precautions for the next 7 days as well.
1. The user should take the last missed tablet as soon as she remembers, even if this means taking two tablets at the same time. She then continues to take tablets at her usual time. The next blister pack must be started as soon as the current blister pack is finished i.e. no gap should be left between packs. The user is unlikely to have a withdrawal bleed until the end of the second pack, but she may experience spotting or breakthrough bleeding on tablet-taking days.
2. The woman may also be advised to discontinue tablet-taking from the current blister pack. She should then have a tablet-free interval of up to 7 days, including the days she missed tablets, and subsequently continue with the next blister pack.
If the woman missed tablets and subsequently has no withdrawal bleed in the first normal tablet-free interval, the possibility of a pregnancy should be considered.
Advice in case of gastro-intestinal disturbances
In case of severe gastro-intestinal disturbances (e.g., vomiting or diarrhoea), absorption may not be complete and additional contraceptive measures should be taken. If vomiting occurs within 3-4 hours after tablet-taking, a new (replacement) tablet should be taken as soon as possible. The new tablet should be taken within 12 hours of the usual time of tablet-taking if possible. If more than 12 hours elapse, the advice concerning missed tablets, as given in section
4.2 “Management of missed tablets”, is applicable. If the woman does not want to change her normal tablet-taking schedule, she has to take the extra tablet(s) from another blister pack.
How to postpone a withdrawal bleed
To delay a period the woman should continue with another blister pack of Acondro without a tablet-free interval. The extension can be carried on for as long as wished until the end of the second pack. During the extension the woman may experience breakthrough-bleeding or spotting. Regular intake of Acondro is then resumed after the usual 7-day tablet-free interval.
To shift her periods to another day of the week than the woman is used to with her current scheme, she can be advised to shorten her forthcoming tablet-free interval by as many days as she likes. The shorter the interval, the higher the risk that she does not have a withdrawal bleed and will experience breakthroughbleeding and spotting during the subsequent pack (just as when delaying a period).
Paediatric population
Acondro is only indicated after menarche. Based on epidemiological data collected on more than 2000 adolescent women aged below 18 years, there are no data indicating that safety and efficacy in this young age group is different from that known in women aged above 18 years.
Method of administration For oral use.
4.3 Contraindications
Combined hormonal contraceptives (CHCs) should not be used in the following
conditions. Should any of the conditions appear for the first time during CHC
use, the product should be stopped immediately.
• Hypersensitivity to the active substances or to any of the excipients listed in section
6.1.
• Presence or risk of venous thromboembolism (VTE)
o Venous thromboembolism - current VTE (on anticoagulants) or history of (e.g. deep venous thrombosis (DVT) or pulmonary embolism (PE))
o Known hereditary or acquired predisposition for venous thromboembolism, such as APC-resistance, (including Factor V Leiden), antithrombin-III-deficiency, protein C deficiency, protein S deficiency
o Major surgery with prolonged immobilisation (see section 4.4) o A high risk of venous thromboembolism due to the presence of multiple risk factors (see section 4.4)
• Presence or risk of arterial thromboembolism (ATE)
o Arterial thromboembolism - current arterial thromboembolism, history of arterial thromboembolism (e.g. myocardial infarction) or prodromal condition (e.g. angina pectoris) o Cerebrovascular disease - current stroke, history of stroke or prodromal condition (e.g. transient ischaemic attack, TIA) o Known hereditary or acquired predisposition for arterial thromboembolism, such as hyperhomocysteinaemia and antiphospholipid-antibodies (anticardiolipin-antibodies, lupus anticoagulant).
o History of migraine with focal neurological symptoms. o A high risk of arterial thromboembolism due to multiple risk factors (see section 4.4) or to the presence of one serious risk factor such as:
■ diabetes mellitus with vascular symptoms
■ severe hypertension
■ severe dyslipoproteinaemia
• Presence or history of severe hepatic disease as long as liver function values
have not returned to normal
• Severe renal insufficiency or acute renal failure
• Presence or history of liver tumours (benign or malignant)
• Known or suspected sex-steroid influenced malignancies (e.g. of the genital
organs or the breasts)
• Undiagnosed vaginal bleeding
4.4 Special warnings and precautions for use
Warnings
If any of the conditions or risk factors mentioned below is present, the suitability of Acondro should be discussed with the woman.
In the event of aggravation, or first appearance of any of these conditions or risk factors, the woman should be advised to contact her doctor to determine whether the use of Acondro should be discontinued.
In case of suspected or confirmed VTE or ATE, CHC use should be discontinued. In case anti-coagulant therapy is started, adequate alternative contraception should be initiated because of the teratogenicity of anticoagulant therapy (coumarins).
Circulatory disorders
Risk of venous thromboembolism (VTE)
The use of any combined hormonal contraceptive (CHC) increases the risk of venous thromboembolism (VTE) compared with no use. Products that contain levonorgestrel, norgestimate or norethisterone are associated with the lowest risk of VTE. Other products such as Acondro may have up to twice this level of risk. The decision to use any product other than one with the lowest VTE risk should be taken only after a discussion with the woman to ensure she understands the risk of VTE with Acondro, how her current risk factors influence this risk, and that her VTE risk is highest in the first ever year of use. There is also some evidence that the risk is increased when a CHC is re-started after a break in use of 4 weeks or more.
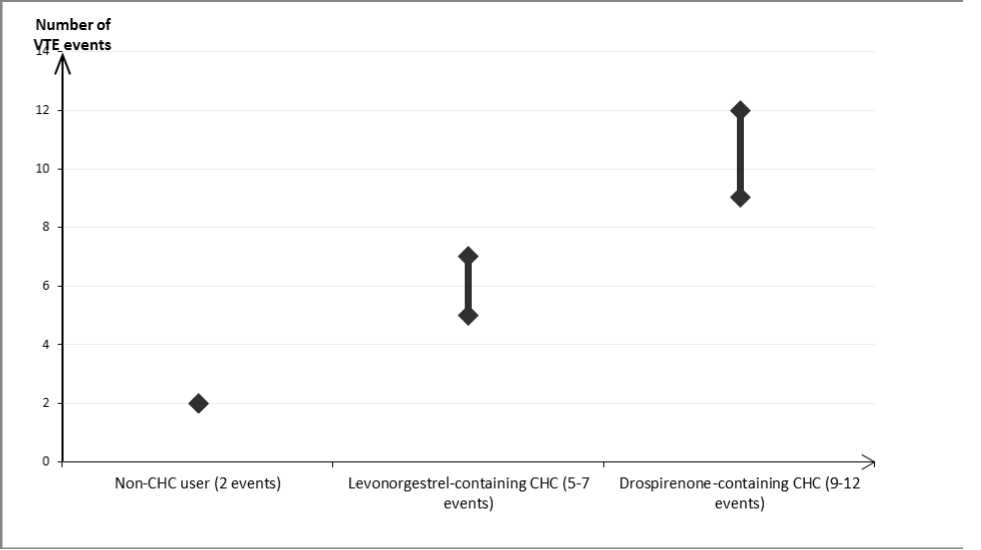
In women who do not use a combined hormonal contraceptive (CHC) and are not pregnant about 2 out of 10,000 will develop a VTE over the period of one year. However, in any individual woman the risk may be far higher, depending on her underlying risk factors (see below).
It is estimated11 2 that out of 10,000 women who use a CHC containing drospirenone, between 9 and 12 women will develop a VTE in one year; this compares with about 6 in women who use a levonorgestrel-containing CHC.
In both cases, the number of VTEs per year is fewer than the number expected during pregnancy or in the postpartum period.
VTE may be fatal in 1-2% of cases.
Extremely rarely, thrombosis has been reported to occur in CHC users in other blood vessels, e.g. hepatic, mesenteric, renal or retinal veins and arteries.
Risk factors for VTE
The risk for venous thromboembolic complications in CHC users may increase substantially in a woman with additional risk factors, particularly if there are multiple risk factors (see table).
Acondro is contraindicated if a woman has multiple risk factors that put her at high risk of venous thrombosis (see section 4.3). If a woman has more than one risk factor, it is possible that the increase in risk is greater than the sum of the
individual factors - in this case her total risk of VTE should be considered. If the balance of benefits and risks is considered to be negative a CHC should not be prescribed (see section 4.3).
Table: Risk factors for VTE
|
Obesity (body mass index over 30 kg/m2) |
Risk increases substantially as BMI rises. |
|
Particularly important to consider if other risk factors also present. | |
|
Prolonged immobilisation, major surgery, any surgery to the legs or pelvis, neurosurgery, or major trauma |
In these situations it is advisable to discontinue use of the pill (in the case of elective surgery at least four weeks in advance) and not resume until two weeks after complete remobilisation. Another method of contraception should be used to avoid unintentional pregnancy. |
|
Antithrombotic treatment should be considered if Acondro has not been discontinued in advance. | |
|
Note: temporary immobilisation including air travel >4 hours can also be a risk factor for VTE, particularly in women with other risk factors | |
|
Positive family history (venous thromboembolism ever in a sibling or parent especially at a relatively early |
If a hereditary predisposition is suspected, the woman should be referred to a specialist for advice before deciding about any CHC use |
|
age e.g. before 50). | |
|
Other medical conditions associated with VTE |
Cancer, systemic lupus erythematosus, haemolytic uraemic syndrome, chronic inflammatory bowel disease (Crohn’s disease or ulcerative colitis) and |
|
sickle cell disease |
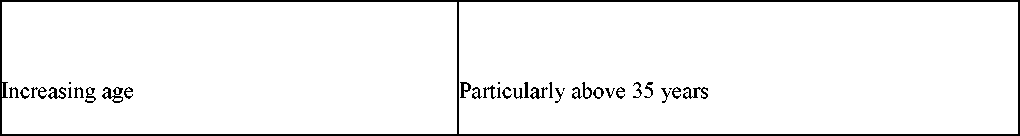
There is no consensus about the possible role of varicose veins and superficial thrombophlebitis in the onset or progression of venous thrombosis.
The increased risk of thromboembolism in pregnancy, and particularly the 6 week period of the puerperium, must be considered (for information on “Pregnancy and lactation” see section 4.6).
Symptoms of VTE (deep vein thrombosis and pulmonary embolism)
In the event of symptoms women should be advised to seek urgent medical attention and to inform the healthcare professional that she is taking a CHC.
Symptoms of deep vein thrombosis (DVT) can include:
- unilateral swelling of the leg and/or foot or along a vein in the leg;
- pain or tenderness in the leg which may be felt only when standing or walking,
- increased warmth in the affected leg; red or discoloured skin on the leg.
Symptoms of pulmonary embolism (PE) can include:
- sudden onset of unexplained shortness of breath or rapid breathing;
- sudden coughing which may be associated with haemoptysis;
- sharp chest pain;
- severe light headedness or dizziness;
- rapid or irregular heartbeat.
Some of these symptoms (e.g. “shortness of breath”, “coughing”) are non-specific and might be misinterpreted as more common or less severe events (e.g. respiratory tract infections).
Other signs of vascular occlusion can include: sudden pain, swelling and slight blue discoloration of an extremity.
If the occlusion occurs in the eye symptoms can range from painless blurring of vision which can progress to loss of vision. Sometimes loss of vision can occur almost immediately.
Risk of arterial thromboembolism (ATE)
Epidemiological studies have associated the use of CHCs with an increased risk for arterial thromboembolism (myocardial infarction) or for cerebrovascular accident (e.g. transient ischaemic attack, stroke). Arterial thromboembolic events may be fatal.
Risk factors for ATE
The risk of arterial thromboembolic complications or of a cerebrovascular accident in CHC users increases in women with risk factors (see table). Acondro is contraindicated if a woman has one serious or multiple risk factors for ATE that puts her at high risk of arterial thrombosis (see section 4.3). If a woman has more than one risk factor, it is possible that the increase in risk is greater than the sum of the individual factors - in this case her total risk should be considered. If the balance of
benefits and risks is considered to be negative a CHC should not be prescribed (see section 4.3).
Table: Risk factors for ATE
|
Risk factor |
Comment |
|
Increasing age |
Particularly above 35 years |
|
Smoking |
Women should be advised not to smoke if they wish to use a CHC. Women over 35 who continue to smoke should be strongly advised to use a different method of contraception. |
|
Hypertension | |
|
Obesity (body mass index over 30 kg/m2) |
Risk increases substantially as BMI increases. Particularly important in women with additional risk factors |
|
Positive family history (arterial thromboembolism ever in a sibling or parent especially at relatively early |
If a hereditary predisposition is suspected, the woman should be referred to a specialist for advice before deciding about any CHC use |
|
age e.g. below 50). | |
|
Migraine |
An increase in frequency or severity of migraine during CHC use (which may be prodromal of a cerebrovascular event) may be a reason for immediate discontinuation |
|
Other medical conditions associated with adverse vascular events |
Diabetes mellitus, hyperhomocysteinaemia, valvular heart disease and atrial fibrillation, dyslipoproteinaemia and systemic lupus erythematosus. |
Symptoms of ATE
In the event of symptoms women should be advised to seek urgent medical attention and to inform the healthcare professional that she is taking a CHC.
Symptoms of a cerebrovascular accident can include:
- sudden numbness or weakness of the face, arm or leg, especially on one side of the body;
- sudden trouble walking, dizziness, loss of balance or coordination;
- sudden confusion, trouble speaking or understanding;
- sudden trouble seeing in one or both eyes;
- sudden, severe or prolonged headache with no known cause;
- loss of consciousness or fainting with or without seizure.
Temporary symptoms suggest the event is a transient ischaemic attack (TIA).
Symptoms of myocardial infarction (MI) can include:
- pain, discomfort, pressure, heaviness, sensation of squeezing or fullness in the chest, arm, or below the breastbone;
- discomfort radiating to the back, jaw, throat, arm, stomach;
- feeling of being full, having indigestion or choking;
- sweating, nausea, vomiting or dizziness;
- extreme weakness, anxiety, or shortness of breath;
- rapid or irregular heartbeats.
• Tumours
An increased risk of cervical cancer in long-term users of COCs (> 5 years) has been reported in some epidemiological studies, but there continues to be controversy about the extent to which this finding is attributable to the confounding effects of sexual behaviour and other factors such as human papilloma virus (HPV).
A meta-analysis from 54 epidemiological studies reported that there is a slightly increased relative risk (RR = 1.24) of having breast cancer diagnosed in women who are currently using COCs. The excess risk gradually disappears during the course of the 10 years after cessation of COC use. Because breast cancer is rare in women under 40 years of age, the excess number of breast cancer diagnoses in current and recent COC users is small in relation to the overall risk of breast cancer. These studies do not provide evidence for causation. The observed pattern of increased risk may be due to an earlier diagnosis of breast cancer in COC users, the biological effects of COCs or a combination of both. The breast cancers diagnosed in ever-users tend to be less advanced clinically than the cancers diagnosed in never-users.
In rare cases, benign liver tumours, and even more rarely, malignant liver tumours have been reported in users of COCs. In isolated cases, these tumours have led to life-threatening intra-abdominal haemorrhages. A hepatic tumour should be considered in the differential diagnosis when severe upper abdominal pain, liver enlargement or signs of intra-abdominal haemorrhage occur in women taking COCs.
With the use of the higher-dosed COCs (50 pg ethinylestradiol) the risk of endometrial and ovarian cancer is reduced. Whether this also applies to lower-dosed COCs remains to be confirmed.
• Other conditions
The progestin component in Acondro is an aldosterone antagonist with potassium sparing properties. In most cases, no increase of potassium levels is to be expected. In a clinical study, however in some patients with mild or moderate renal impairment and concomitant use of potassium-sparing medicinal products serum potassium levels slightly, but not significantly, increased during drospirenone intake. Therefore, it is recommended to check serum potassium during the first treatment cycle in patients presenting with renal insufficiency and a pretreatment serum potassium in the upper reference range, and particularly during concomitant use of potassium sparing medicinal products. See also section 4.5.
Women with hypertriglyceridemia, or a family history thereof, may be at an increased risk of pancreatitis when using COCs.
Although small increases in blood pressure have been reported in many women taking COCs, clinically relevant increases are rare. Only in these rare cases an immediate discontinuation of COC use is justified. If, during the use of a COC in preexisting hypertension, constantly elevated blood pressure values or a significant increase in blood pressure do not respond adequately to antihypertensive treatment, the COC must be withdrawn. Where considered appropriate, COC use may be resumed if normotensive values can be achieved with antihypertensive therapy.
The following conditions have been reported to occur or deteriorate with both pregnancy and COC use, but the evidence of an association with COC use is inconclusive: jaundice and/or pruritus related to cholestasis; gallstones; porphyria; systemic lupus erythematosus; haemolytic uraemic syndrome; Sydenham's chorea; herpes gestationis; otosclerosis-related hearing loss.
In women with hereditary angioedema exogenous oestrogens may induce or exacerbate symptoms of angioedema.
Acute or chronic disturbances of liver function may necessitate the discontinuation of COC use until markers of liver function return to normal. Recurrence of cholestatic jaundice and/or cholestasis-related pruritus which previously occurred during pregnancy or during previous use of sex steroids necessitates the discontinuation of COCs.
Although COCs may have an effect on peripheral insulin resistance and glucose tolerance, there is no evidence for a need to alter the therapeutic regimen in diabetics using low-dose COCs (containing < 0.05 mg ethinylestradiol). However, diabetic women should be carefully observed, particularly in the early stage of COC use.
Worsening of endogenous depression, of epilepsy, of Crohn's disease and of ulcerative colitis has been reported during COC use.
Chloasma may occasionally occur, especially in women with a history of chloasma gravidarum. Women with a tendency to chloasma should avoid exposure to the sun or ultraviolet radiation whilst taking COCs.
Medical examination/consultation
Prior to the initiation or reinstitution of Acondro a complete medical history (including family history) should be taken and pregnancy must be ruled out. Blood pressure should be measured and a physical examination should be performed, guided by the contra-indications (see section 4.3) and warnings (see section 4.4). It is important to draw a woman’s attention to the information on venous and arterial thrombosis, including the risk of Acondro compared with other CHCs, the symptoms of VTE and ATE, the known risk factors and what to do in the event of a suspected thrombosis.The woman should also be instructed to carefully read the user leaflet and to adhere to the advice given. The frequency and nature of examinations should be based on established practice guidelines and be adapted to the individual woman.
Women should be advised that hormonal contraceptives do not protect against HIV infections (AIDS) and other sexually transmitted diseases.
Reduced efficacy
The efficacy of COCs may be reduced in the event of e.g. missed tablets (see section 4.2), gastro-intestinal disturbances (see section 4.2) or concomitant medication (see section 4.5).
Reduced cycle control
With all COCs, irregular bleeding (spotting or breakthrough bleeding) may occur, especially during the first months of use. Therefore, the evaluation of any irregular bleeding is only meaningful after an adaptation interval of about three cycles.
If bleeding irregularities persist or occur after previously regular cycles, then nonhormonal causes should be considered and adequate diagnostic measures are indicated to exclude malignancy or pregnancy. These may include curettage.
In some women withdrawal bleeding may not occur during the tablet-free interval. If the COC has been taken according to the directions described in section 4.2, it is unlikely that the woman is pregnant. However, if the COC has not been taken according to these directions prior to the first missed withdrawal bleed or if two withdrawal bleeds are missed, pregnancy must be ruled out before COC use is continued.
This medicinal product contains 62 mg lactose per tablet. Patients with rare hereditary problems of galactose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption who are on a lactose-free diet should take this amount into consideration.
4.5 Interaction with other medicinal products and other forms of interaction
Note: The prescribing information of concomitant medications should be consulted to identify potential interactions.
• Effects of other medicinal products on Acondro
Interactions can occur with drugs that induce microsomal enzymes which can result in increased clearance of sex hormones and which may lead to breakthrough bleeding and/or contraceptive failure.
Management
Enzyme induction can already be observed after a few days of treatment. Maximal enzyme induction is generally seen within a few weeks. After the cessation of drug therapy enzyme induction may be sustained for about 4 weeks.
Short-term treatment
Women on treatment with enzyme-inducing drugs should temporarily use a barrier method or another method of contraception in addition to the COC. The barrier method must be used during the whole time of the concomitant drug therapy and for 28 days after its discontinuation. If the drug therapy runs beyond the end of the tablets in the COC pack, the next COC pack should be started right after the previous one without the usual tablet-free interval.
Long-term treatment
In women on long-term treatment with hepatic enzyme-inducing active substances, another reliable, non-hormonal, method of contraception is recommended.
The following interactions have been reported in the literature.
Substances increasing the clearance of COCs (diminished efficacy of COCs by enzyme-induction), e.g.:
Barbiturates, bosentan, carbamazepine, phenytoin, primidone, rifampicin, and HIV medication ritonavir, nevirapine and efavirenz and possibly also felbamate, griseofulvin, oxcarbazepine, topiramate and products containing the herbal remedy St. John's wort (hypericum perforatum).
Substances with variable effects on the clearance of COCs:
When co-administered with COCs many combinations of HIV protease inhibitors and non-nucleoside reverse transcriptase inhibitors, including combinations with HCV inhibitors can increase or decrease plasma concentration of oestrogen or progestins. The net effect of these changes may be clinically relevant in some cases.
Therefore, the prescribing information of concomitant HIV/HCV medications should be consulted to identify potential interactions and any related recommendations. In case of any doubt, an additional barrier contraceptive method should be used by women on protease inhibitor or non-nucleoside reverse transcriptase inhibitor therapy.
Substances decreasing the clearance of COCs (enzyme inhibitors):
The clinical relevance of potential interactions with enzyme inhibitors remains unknown.
Concomitant administration of strong CYP3A4 inhibitors can increase plasma concentrations of the oestrogen or the progestin or both.
In a multiple dose study with a drospirenone (3 mg/day) / ethinylestradiol (0.02 mg/day) combination, coadministration of the strong CYP3A4 inhibitor ketoconazole for 10 days increased the AUC (0-24h) of drospirenone and ethinylestradiol 2.7-fold and 1.4-fold, respectively.
Etoricoxib doses of 60 to 120 mg/day have been shown to increase plasma concentrations of ethinylestradiol 1.4 to 1.6-fold, respectively when taken concomitantly with a combined hormonal contraceptive containing 0.035 mg ethinylestradiol.
• Effects of Acondro on other medicinal products
Oral contraceptives may affect the metabolism of certain other active substances. Accordingly, plasma and tissue concentrations may either increase (e.g. ciclosporin) or decrease (e.g. lamotrigine).
Based on in vivo interaction studies in female volunteers using omeprazole, simvastatin or midazolam as marker substrate, a clinically relevant interaction of drospirenone at doses of 3 mg with the cytochrome P450 mediated metabolism of other active substances is unlikely.
Clinical data suggests that ethinylestradiol is inhibiting the clearance of CYP1A2 substrates leading to a weak (e.g. theophylline) or moderate (e.g. tizanidine) increase in their plasma concentration.
• Other forms of interaction
In patients without renal insufficiency, the concomitant use of drospirenone and ACE-inhibitors or NSAIDs did not show a significant effect on serum potassium. Nevertheless, concomitant use of Acondro with aldosterone antagonists or potassiumsparing diuretics has not been studied. In this case, serum potassium should be tested during the first treatment cycle. See also section 4.4.
• Laboratory tests
The use of contraceptive steroids may influence the results of certain laboratory tests, including biochemical parameters of liver, thyroid, adrenal and renal function, plasma levels of (carrier) proteins, e.g. corticosteroid-binding globulin and lipid/lipoprotein fractions, parameters of carbohydrate metabolism and parameters of coagulation and fibrinolysis. Changes generally remain within the normal laboratory range. Drospirenone causes an increase in plasma renin activity and plasma aldosterone induced by its mild antimineralocorticoid activity.
4.6 Fertility, pregnancy and lactation
Pregnancy
Acondro is not indicated during pregnancy.
If pregnancy occurs during use of Acondro, the preparation should be withdrawn immediately. Extensive epidemiological studies have revealed neither an increased risk of birth defects in children born to women who used COCs prior to pregnancy, nor a teratogenic effect when COCs were taken inadvertently during pregnancy.
Animal studies have shown undesirable effects during pregnancy and lactation (see section 5.3). Based on these animal data, undesirable effects due to hormonal action of the active compounds cannot be excluded. However, general experience with COCs during pregnancy did not provide evidence for an actual undesirable effect in humans.
The available data regarding the use of Acondro during pregnancy are too limited to permit conclusions concerning negative effects of Acondro on pregnancy, health of the foetus or neonate. To date, no relevant epidemiological data are available.
The increased risk of VTE during the postpartum period should be considered when re-starting Acondro (see sections 4.2 and 4.4).
Breast-feeding
Lactation may be influenced by COCs as they may reduce the quantity and change the composition of breast milk. Therefore, the use of COCs should generally not be recommended until the breast-feeding mother has completely weaned her child. Small amounts of the contraceptive steroids and/or their metabolites may be excreted with the milk during COC use. These amounts may affect the child.
4.7 Effects on ability to drive and use machines
No studies on the effects on the ability to drive and use machines have been performed. No effects on ability to drive and use machines have been observed in users of COCs.
4.8 Undesirable effects
For serious undesirable effects in COC users see section 4.4
The following adverse reactions have been reported during use of Acondro:
|
System Organ Class |
Fret |
uency of adverse reactions | |
|
Common > 1/100 to <1/10 |
Uncommon > 1/1,000 to <1/100 |
Rare > 1/10,000 to <1/1,000 | |
|
Immune system disorders |
Hypersensitivity Asthma | ||
|
Psychiatric disorders |
Depressive mood |
Libido increased, Libido decreased | |
|
Nervous system disorders |
Headache | ||
|
Ear and labyrinth disorders |
Hypoacusis | ||
|
Vascular disorders |
Migraine |
Hypertension Hypotension |
Venous thromboembolism (VTE) Arterial thromboembolism (ATE) |
|
Gastrointestinal disorders |
Nausea |
Vomiting Diarrhoea | |
|
Skin and subcutaneous tissue disorders |
Acne Eczema Pruritus Alopecia |
Erythema nodosum Erythema multiforme | |
|
Reproductive system and breast disorders |
Menstrual disorders Intermenstrual bleeding Breast pain Breast tenderness Vaginal discharge Vulvovaginal candidiasis |
Breast enlargement Vaginal infection |
Breast secretion |
|
General disorders and administration site conditions |
Fluid retention Weight increased Weight decreased | ||
Description of selected adverse reactions
An increased risk of arterial and venous thrombotic and thrombo-embolic events, including myocardial infarction, stroke, transient ischemic attacks, venous thrombosis and pulmonary embolism has been observed in women using CHCs, which are discussed in more detail in section 4.4.
The following serious adverse events have been reported in women using COCs, which are discussed in section 4.4:
• Venous thromboembolic disorders;
• Arterial thromboembolic disorders;
• Hypertension;
• Liver tumours;
• Occurrence or deterioration of conditions for which association with COC use is not conclusive: Crohn’s disease, ulcerative colitis, epilepsy, uterine myoma, porphyria, systemic lupus erythematosus, herpes gestationis, Sydenham's chorea, haemolytic uraemic syndrome, cholestatic jaundice;
• Chloasma;
• Acute or chronic disturbances of liver function may necessitate the discontinuation of COC use until markers of liver function return to normal.
• In women with hereditary angioedema exogenous oestrogens may induce or exacerbate symptoms of angioedema
The frequency of diagnosis of breast cancer is very slightly increased among COC users. As breast cancer is rare in women under 40 years of age the excess number is small in relation to the overall risk of breast cancer. Causation with COC use is unknown. For further information, see sections 4.3 and 4.4.
Interactions
Breakthrough bleeding and/or contraceptive failure may result from interactions of other drugs (enzyme inducers) with oral contraceptives (see section 4.5).
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Yellow Card Scheme at: www.mhra.gov.uk/yellowcard
4.9 Overdose
There has not yet been any experience of overdose with Acondro. On the basis of general experience with combined oral contraceptives, symptoms that may possibly occur in this case are: nausea, vomiting and, in young girls, slight vaginal bleeding. There are no antidotes and further treatment should be symptomatic.
5. PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Progestogens and oestrogens, fixed combinations, ATC code: G03AA12
Pearl Index for method failure: 0.09 (upper two-sided 95% confidence limit: 0.32) Overall Pearl Index (method failure + patient failure): 0.57 (upper two-sided 95 % confidence limit: 0.90).
Mechanism of action
The contraceptive effect of Acondro is based on the interaction of various factors, the most important of which are seen as the inhibition of ovulation and the changes in the endometrium.
Pharmacodynamic effects
Acondro is a combined oral contraceptive with ethinylestradiol and the progestogen drospirenone. In a therapeutic dosage, drospirenone also possesses antiandrogenic and mild antimineralocorticoid properties. It has no oestrogenic, glucocorticoid and antiglucocorticoid activity. This gives drospirenone a pharmacological profile closely resembling the natural hormone progesterone.
There are indications from clinical studies that the mild antimineralocorticoid properties of Acondro result in a mild antimineralocorticoid effect.
5.2. .Pharmacokinetic properties Drospirenone
Absorption
Orally administered drospirenone is rapidly and almost completely absorbed. Maximum concentrations of the active substance in serum of about 38 ng/ml are reached at about 1 - 2 h after single ingestion. Bioavailability is between 76 and 85%. Concomitant ingestion of food has no influence on the bioavailability of drospirenone.
Distribution
After oral administration, serum drospirenone levels decrease with a terminal half-life of 31 h. Drospirenone is bound to serum albumin and does not bind to sex hormone binding globulin (SHBG) or corticoid binding globulin (CBG). Only 3 - 5 % of the total serum concentrations of the active substance are present as free steroid. The ethinylestradiol-induced increase in SHBG does not influence the serum protein binding of drospirenone. The mean apparent volume of distribution of drospirenone is 3.7 ± 1.2 l/kg.
Biotransformation
Drospirenone is extensively metabolised after oral administration. The major metabolites in the plasma are the acid form of drospirenone, generated by opening of the lactone ring, and the 4,5-dihydro-drospirenone-3-sulfate formed by reduction and subsequent sulfatation. Drospirenone is also subject to oxidative metabolism catalysed by CYP3A4.
In vitro, drospirenone is capable to inhibit weakly to moderately the cytochrome P450 enzymes CYP 1A1, CYP 2C9, CYP 2C19 and CYP3A4.
Elimination
The metabolic clearance rate of drospirenone in serum is 1.5 ± 0.2 ml/min/kg. Drospirenone is excreted only in trace amounts in unchanged form. The metabolites of drospirenone are excreted with the faeces and urine at an excretion ratio of about
1.2 to 1.4. The half-life of metabolite excretion with the urine and faeces is about 40h.
Steady-State Conditions
During a treatment cycle, maximum steady-state concentrations of drospirenone in serum of about 70 ng/ml are reached after about 8 days of treatment. Serum drospirenone levels accumulated by a factor of about 3 as a consequence of the ratio of terminal half-life and dosing interval.
Special Populations Effect of renal impairment
Steady-state serum drospirenone levels in women with mild renal impairment (creatinine clearance CLcr, 50-80 mL/min) were comparable to those of women with normal renal function. The serum drospirenone levels were on average 37 % higher in women with moderate renal impairment (CLcr, 30 - 50 mL/min) compared to those in women with normal renal function. Drospirenone treatment was also well tolerated by women with mild and moderate renal impairment. Drospirenone treatment did not show any clinically significant effect on serum potassium concentration.
Effect of hepatic impairment
In a single dose study, oral clearance (CL/F) was decreased approximately 50 % in volunteers with moderate hepatic impairment as compared to those with normal liver function. The observed decline in drospirenone clearance in volunteers with moderate hepatic impairment did not translate into any apparent difference in terms of serum potassium concentrations. Even in the presence of diabetes and concomitant treatment with spironolactone (two factors that can predispose a patient to hyperkalemia) an increase in serum potassium concentrations above the upper limit of the normal range was not observed. It can be concluded that drospirenone is well tolerated in patients with mild or moderate hepatic impairment (Child-Pugh B).
Ethnic groups
No clinically relevant differences in the pharmacokinetics of drospirenone or ethinylestradiol between Japanese and Caucasian women have been observed.
Ethinylestradiol
Absorption
Ethinylestradiol is rapidly and completely absorbed after ingestion. After administration of 30 pg, peak plasma concentrations of 100 pg/ml are reached 1-2 hours after ingestion. Ethinylestradiol undergoes an extensive first-pass effect, which displays great inter-individual variation. The absolute bioavailability is approx. 45 %.
Distribution
Ethinylestradiol has an apparent volume of distribution of 5 l/kg and binding to plasma proteins is approx. 98 %. Ethinylestradiol induces the hepatic synthesis of SHBG and CBG. During treatment with 30 pg ethinylestradiol the plasma concentration of SHBG increases from 70 to about 350 nmol/l.
Ethinylestradiol passes in small amounts into breast milk (0.02 % of the dose).
Biotransformation
Ethinylestradiol is subject to significant gut and hepatic first-pass metabolism. Ethinylestradiol is primarily metabolised by aromatic hydroxylation but a wide variety of hydroxylated and methylated metabolites are formed, and these are present as free metabolites and as conjugates with glucuronides and sulfate. The metabolic clearance rate of ethinylestradiol is about 5 ml/min/kg.
In vitro, ethinylestradiol is a reversible inhibitor of CYP2C19, CYP1A1 and CYP1A2 as well as a mechanism based inhibitor of CYP3A4/5, CYP2C8, and CYP2J2.
Elimination
Ethinylestradiol is not excreted in unchanged form to any significant extent. The metabolites of ethinylestradiol are excreted at a urinary to biliary ratio of 4:6. The half-life of metabolite excretion is about 1 day. The elimination half-life is 20 hours.
Steady-state conditions
Steady-state conditions are reached during the second half of a treatment cycle and serum levels of ethinylestradiol accumulate by a factor of about 1.4 to 2.1.
5.3 Preclinical safety data
In laboratory animals, the effects of drospirenone and ethinylestradiol were confined to those associated with the recognised pharmacological action. In particular, reproduction toxicity studies revealed embryotoxic and fetotoxic effects in animals which are considered as species specific. At exposures exceeding those in users of Acondro, effects on sexual differentiation were observed in rat foetuses but not in monkeys.
6. PHARMACEUTICAL PARTICULARS
6.1 List of excipients
Tablet core:
Lactose monohydrate Maize starch
Pregelatinised starch (maize)
Crospovidone type A Crospovidone type B Povidone (E1201)
Polysorbate 80 (E433)
Magnesium stearate (E470b)
Coating:
Polyvinyl alcohol partial hydrolysed Titanium dioxide (E171)
Macrogol Talc (E553b)
Yellow iron oxide (E172)
6.2 Incompatibilities
Not applicable.
6.3 Shelf life
3 years.
6.4 Special precautions for storage
Store below 30°C.
6.5 Nature and contents of container
Transparent PVC/PVDC film in aluminium push-through foil blister packs Pack sizes:
1 x 21 film-coated tablets
2 x 21 film-coated tablets
3 x 21 film-coated tablets 6 x 21 film-coated tablets 13 x 21 film-coated tablets
Not all pack sizes may be marketed.
6.6 Special precautions for disposal
No special requirements.
7 MARKETING AUTHORISATION HOLDER
Mylan, Potters Bar, Hertfordshire, EN6 1TL, United Kingdom
8 MARKETING AUTHORISATION NUMBER(S)
9 DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
14/05/2013
10 DATE OF REVISION OF THE TEXT
08/08/2015
These incidences were estimated from the totality of the epidemiological study data, using relative risks for the different products compared with levonorgestrel-containing CHCs.
Mid-point of range of 5-7 per 10,000 WY, based on a relative risk for CHCs containing levonorgestrel versus non-use of approximately 2.3 to 3.6