Azzalure 10 Speywood Units/0.05Ml Powder For Solution For Injection
SUMMARY OF PRODUCT CHARACTERISTICS
1 NAME OF THE MEDICINAL PRODUCT
Azzalure, 10 Speywood units/0.05ml, powder for solution for injection
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
Botulinum toxin type A * 10 Speywood units **/0.05ml of reconstituted solution Vial of 125 units
* Clostridium botulinum toxin A haemagglutinin complex
**One Speywood unit (U) is defined as the median lethal peritoneal dose in mice (LD50).
The Speywood units of Azzalure are specific to the preparation and are not interchangeable with other preparations of botulinum toxin
For a full list of excipients see Section 6.1.
3 PHARMACEUTICAL FORM
Powder for solution for injection.
The powder is white.
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
Azzalure is indicated for the temporary improvement in the appearance of moderate to severe glabellar lines (vertical lines between the eyebrows) seen at frown, in adult patients under 65 years, when the severity of these lines has an important psychological impact on the patient.
4.2 Posology and method of administration
Posology:
Botulinum toxin units are different depending on the medicinal products. The Speywood units of Azzalure are specific to the preparation and are not interchangeable with other preparations of botulinum toxin.
Paediatric Population
The safety and effectiveness of Azzalure in individuals under 18 years of age have not been demonstrated.
Method of administration (see section 6.6):
Azzalure should only be administered by physicians with appropriate qualifications and expertise in this treatment and having the required equipment.
Once reconstituted, Azzalure should only be used to treat a single patient, during a single session.
Prior to injection, the product should be reconstituted, instructions for which are given in Section 6.6.
Remove any make-up and disinfect the skin with a local antiseptic.
Intramuscular injections should be performed at right angles to the skin using a sterile 29-30 gauge needle.
Administration instructions:
The recommended dose is 50 Speywood units (0.25 ml of reconstituted solution) of Azzalure to be divided into 5 injection sites, 10 Speywood units (0.05 ml of reconstituted solution) are to be administered intramuscularly into each of the 5 sites: 2 injections into each corrugator muscle and one into the procerus muscle near the nasofrontal angle as shown below:
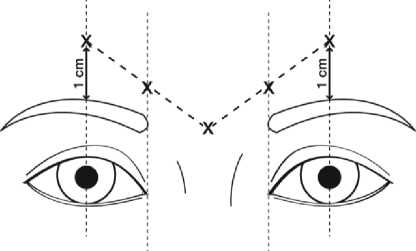
The anatomical landmarks can be more readily identified if observed and palpated at maximal frown. Before injection, place the thumb or index finger firmly below the orbital rim in order to prevent extravasation below the orbital rim. The needle should be pointed upward and medially during the injection. In order to reduce the risk of ptosis, avoid injections near the levator palpebrae superioris muscle, particularly in patients with larger brow-depressor complexes (depressor supercilii). Injections in the corrugator muscle must be made into the central part of that muscle, at least 1 cm above the orbital rim.
The treatment interval depends on the individual patient’s response after assessment. In clinical studies, an optimal effect was demonstrated for up to 4 months after injection. Some patients were still responders at 5 months (see section 5.1). Treatment interval should not be more frequent than every three months.
In the event of treatment failure or diminished effect following repeat injections, alternative treatment methods should be employed. In case of treatment failure after the first treatment session, the following approaches may be considered:
• Analysis of the causes of failure, e.g. incorrect muscles injected, injection technique, and formation of toxin-neutralising antibodies;
• Re-evaluation of the relevance of treatment with botulinum toxin A
4.3 Contraindications
- Hypersensitivity to the active substance or to any of the excipients listed in section 6.1;
- In the presence of infection at the proposed injection sites;
- In the presence of myasthenia gravis, Eaton Lambert Syndrome or Amyotrophic lateral sclerosis.
4.4 Special warnings and precautions for use
Azzalure should be used with caution in patients with a risk of, or clinical evidence of, marked defective neuro-muscular transmission. Such patients may have an increased sensitivity to agents such as Azzalure, which may result in excessive muscle weakness.
Adverse reactions possibly related to the distribution of the effects of the toxin to sites remote from the site of administration have been reported very rarely with botulinum toxin. Patients treated with therapeutic doses may experience exaggerated muscle weakness.
Injection of Azzalure is not recommended in patients with a history of dysphagia and aspiration.
Patients or care-givers should be advised to seek immediate medical care if swallowing, speech or respiratory difficulties arise.
The recommended dose and frequency of administration for Azzalure must not be exceeded.
It is essential to study the patient’s facial anatomy prior to administering Azzalure. Facial asymmetry, ptosis, excessive dermatochalasis, scarring and any alterations to this anatomy, as a result of previous surgical interventions should be taken into consideration.
Caution should be taken when Azzalure is used in the presence of inflammation at the proposed injection site(s) or when the targeted muscle shows excessive weakness or atrophy.
As with all intramuscular injections, Azzalure treatment is not recommended in patients who have a prolonged bleeding time.
Injections at more frequent intervals or at higher doses can increase the risk of antibody formation to botulinum toxin. Clinically, the formation of neutralising antibodies may reduce the effectiveness of subsequent treatment.
The effect of administering different botulinum neurotoxins during the course of treatment with Azzalure is unknown and must be avoided.
It is mandatory that Azzalure is used for one single patient treatment only during a single session. The excess of unused product must be disposed of as detailed in section 6.6. Particular precautions should be taken for product preparation and administration as well as for the inactivation and disposal of the remaining unused solution (see Section 6.6).
4.5 Interaction with other medicinal products and other forms of interaction
Concomitant treatment of Azzalure and aminoglycosides or other agents interfering with neuromuscular transmission (e.g., curare-like agents) should only be used with caution since the effect of botulinum toxin type A may be potentiated.
No interaction studies have been performed. No other interactions of clinical significance have been reported.
4.6 Pregnancy and lactation
Pregnancy
Azzalure should not be used during pregnancy unless clearly necessary. There are no adequate data from the use of botulinum toxin type A in pregnant women. Studies in animals have shown reproductive toxicity at high doses (see Section 5.3). The potential risk for humans is unknown.
Breastfeeding
There is no information on whether Azzalure is excreted in human milk. The use of Azzalure during lactation cannot be recommended.
4.7 Effects on ability to drive and use machines
There is a potential risk of localised muscle weakness or visual disturbances linked with the use of this medicinal product which may temporarily impair the ability to drive or operate machinery.
4.8 Undesirable effects
More than 1900 patients were exposed to Azzalure in the different clinical trials.
In pivotal clinical studies, over 1500 patients with moderate to severe glabellar lines have been treated at the recommended dose of 50 Units in double-blind placebo-controlled and long-term open-label studies.
In pivotal double-blind placebo-controlled single dose studies, 22.3% of patients treated at the recommended Azzalure dose (50U) and 16.6% of patients treated with placebo, experienced a reaction that was related to treatment, injection technique or both. In the long-term open-label dose Phase III study in which patients received multiple injection cycles, 26% of patients experienced at least one related reaction after the first injection. The incidence of treatment/injection technique related reactions decreased over repeat cycles.
The most frequently occurring related reactions are headache and injection site reactions. In general, treatment/injection technique related reactions occur within the first week following injection and are transient. Most of these reactions reported were of mild to moderate severity and were reversible.
The frequency of undesirable effects is classified as follows:
Very common (> 1/10); common (> 1/100 to < 1/10); uncommon (> 1/1,000 to <1/100); rare (> 1/10,000 to < 1/1,000); very rare (< 1/10,000); not known (cannot be estimated from the available data).
|
Nervous system disorders |
Very Common Headache Common Facial paresis (predominantly describes brow paresis) Uncommon Dizziness |
|
Eye disorders |
Common Asthenopia, Ptosis, Eyelid oedema, Lacrimation increase, Dry eye, Muscle twitching (twitching of muscles around the eyes) Uncommon Visual disturbances, Vision blurred, Diplopia, Eye movement disorder |
|
Skin and subcutaneous tissue disorders |
Uncommon Pruritus, Rash Rare Urticaria |
|
General disorders and administration site conditions |
Very Common Injection site reactions (e.g. erythema, oedema, irritation, rash, pruritus, paraesthesia, pain, discomfort, stinging and bruising) |
|
Immune system disorders |
Uncommon Hypersensitivity |
Adverse effects resulting from distribution of the effects of the toxin to sites remote from the site of injection have been very rarely reported with botulinum toxin (excessive muscle weakness, dysphagia, aspiration pneumonia with fatal outcome in some cases) (see section 4.4).
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Yellow Card Scheme at: www.mhra.gov.uk/yellowcard
4.9 Overdose
There were no cases of overdose during clinical studies.
Excessive doses of botulinum toxin may be expected to produce neuromuscular weakness with a variety of symptoms. Respiratory support may be required where excessive doses cause paralysis of respiratory muscles. In the event of overdose the patient should be medically monitored for symptoms of excessive muscle weakness or muscle paralysis. Symptomatic treatment should be instigated if necessary.
Symptoms of overdose may not present immediately following injection.
Admission to hospital should be considered in patients presenting symptoms of botulinum toxin A poisoning (e.g. a combination of muscle weakness, ptosis, diplopia, swallowing and speech disorders, or paresis of the respiratory muscles).
5 PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Other muscle relaxants, peripherally acting agents ATC code: M03AX01
The primary pharmacodynamic effect of Clostridium botulinum toxin type A is due to chemical denervation of the treated muscle resulting in a measurable decrease of the compound muscle action potential, causing a localized reduction of, or paralysis in, muscle activity.
Clinical data
During the clinical development of Azzalure, more than 2600 patients were included in the different clinical trials.
In clinical studies, 1907 patients with moderate to severe glabellar lines have been treated at the recommended dose of 50 Speywood Units. Of these, 305 were treated with 50U in two pivotal Phase III double-blind placebo-controlled studies and 1200 treated with 50U in a long-term open-label repeated dose Phase III study. The remaining patients were treated in supportive and dose-ranging studies.
The median time to onset of response was 2 to 3 days following treatment, with the maximum effect observed at day thirty. In both pivotal placebo-controlled phase III studies, Azzalure injections significantly reduced the severity of glabellar lines for up
to 4 months. The effect was still significant after 5 months in one of the two pivotal studies.
Thirty days following injection, the assessment of the investigators showed that 90% (273/305) of patients had responded to treatment (exhibited no or mild glabellar lines at maximum frown), compared to 3% (4/153) placebo-treated patients. Five months after injection, 17% (32/190) of patients treated with Azzalure were still responding to treatment compared to 1% (1/92) of placebo treated patients in the concerned study. The patients own assessment at maximum frown after thirty days gave a response rate of 82% (251/305) for those treated with Azzalure and 6% (9/153) for those treated with placebo. The proportion of patients exhibiting a two-grade improvement according to the investigator assessment at maximum frown, was 77% (79/103) in the one pivotal Phase III study where this was assessed.
A subset of 177 patients had moderate or severe glabellar lines at rest prior to treatment. Assessment by investigators of this population, thirty days after treatment, showed that 71% (125/177) of Azzalure-treated patients were considered responders versus 10% (8/78) of placebo-treated patients.
The long-term repeat dose open label study showed that the median time to onset of response of 3 days was maintained across repeated dose cycles. The responder rate at maximum frown as determined by the investigator at day 30 was maintained over repeated cycles (ranging between 80% and 91% over the 5 cycles). The responder rate at rest over repeated dose cycles was also consistent with the single dose studies, with 56% to 74% of Azzalure-treated patients considered by investigators to be responders thirty days after treatment.
None of the clinical endpoints included an objective evaluation of the psychological impact.
5.2 Pharmacokinetic properties
Azzalure is not expected to be present in the peripheral blood at measurable levels following IM injection at the recommended dose. Therefore pharmacokinetic studies have not been performed with Azzalure.
5.3 Preclinical safety data
In reproductive studies in rats and rabbits, severe maternal toxicity associated with implantation loses was observed at high doses. At doses corresponding to 60 to 100 times the human recommended dose (50U) in rabbits and rats respectively, no embryofetal toxicity was observed. No teratogenic effects were observed in these species. In rats, fertility of the males and females was decreased due to reduced mating secondary to muscle paralysis at high doses.
In a chronic toxicity study performed in rats, there was no indication of systemic toxicity at doses corresponding to 75 times the human recommended dose (50U) divided equally between the right and left gluteus muscles.
Studies on acute toxicity, chronic toxicity and local tolerance at the injection site did not show unusual adverse local or systemic effects at clinically relevant dose levels.
6 PHARMACEUTICAL PARTICULARS
6.1 List of excipients
Human Albumin 200g/L.
Lactose Monohydrate.
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those mentioned in section 6.6.
6.3 Shelf life 2 years.
Reconstituted solution:
Chemical and physical in-use stability has been demonstrated for 24 hours between 2 - 8°C. From a microbiological point of view, unless the method of reconstituting precludes the risks of microbial contamination, the product should be used immediately.
If not used immediately, in-use storage times and conditions are the responsibility of the user.
6.4 Special precautions for storage
Store in a refrigerator (2°C - 8°C).
Do not freeze.
For storage of the reconstituted medicinal product see Section 6.3.
Nature and contents of container
6.5
125 Speywood Units in a powder in a vial (Type I glass), with a stopper (halobutyl) and seal (aluminium).
Pack size of 1 or 2 vial(s).
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
The instructions for use, handling and disposal should be strictly followed.
Reconstitution should be performed in accordance with good practice rules, particularly in the respect of asepsis.
Azzalure has to be reconstituted with a sodium chloride 9 mg/ml (0.9%) solution for injection. As per the dilution table below, the requested amount of sodium chloride 9 mg/ml (0.9%) solution for injection has to be drawn up into a syringe in order to obtain a reconstituted clear solution at a concentration of 10 U/0.05 ml;
|
Amount of solvent added (0.9% sodium chloride solution) to a 125 U vial |
Resulting dose (Units per 0.05 ml) |
|
0.63 ml |
10 U |
The accurate measurement of 0.63ml can be achieved using 1ml syringes, graduated in 0.1 ml and 0.01 ml increments.
RECOMMENDATIONS FOR THE DISPOSAL OF CONTAMINATED MATERIALS
Immediately after use and prior to disposal, unused reconstituted Azzalure (in the vial or in the syringe) should be inactivated with 2ml of dilute sodium hypochlorite solution at 0.55 or 1% (Dakin’s solution).
Used vials, syringes and materials should not be emptied and must be discarded into appropriate containers and disposed of in accordance with local requirements.
RECOMMENDATIONS SHOULD ANY INCIDENT OCCUR DURING THE HANDLING OF BOTULINUM TOXIN
• Any spills of the product must be wiped up: either using absorbent material impregnated with a solution of sodium hypochlorite (bleach) in case of the powder, or with dry, absorbent material in case of reconstituted product.
• The contaminated surfaces should be cleaned using absorbent material impregnated with a solution of sodium hypochlorite (bleach), then dried.
• If a vial is broken, proceed as mentioned above by carefully collecting the pieces of broken glass and wiping up the product, avoiding any cuts to the skin.
• If the product comes into contact with the skin, wash the affected area with a solution of sodium hypochlorite (bleach) then rinse abundantly with water.
• If product enters into contact with the eyes, rinse thoroughly with plenty of water or with an ophthalmic eyewash solution.
• If product enters into contact with a wound, cut or broken skin, rinse thoroughly with plenty of water and take the appropriate medical steps according to the dose injected.
These instructions for use handling and disposal should be strictly followed.
7 MARKETING AUTHORISATION HOLDER
Ipsen Limited 190 Bath Road Slough SL1 3XE United Kingdom
8 MARKETING AUTHORISATION NUMBER(S)
PL 06958/0031
9 DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
28/01/2014
10 DATE OF REVISION OF THE TEXT
29/10/2015