Botox 200 Allergan Units Powder For Solution For Injection
SUMMARY OF PRODUCT CHARACTERISTICS
1 NAME OF THE MEDICINAL PRODUCT
BOTOX
200 Allergan Units
Powder for solution for injection
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
Botulinum toxin* type A, 200 Allergan Units/vial. from Clostridium botulinum
Botulinum toxin units are not interchangeable from one product to another.
For a full list of excipients, see section 6.1.
3 PHARMACEUTICAL FORM
Powder for solution for injection.
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
BOTOX is indicated for:
Neurologic disorders:
• treatment of focal spasticity, including:
> dynamic equinus foot deformity due to spasticity in ambulant paediatric cerebral palsy patients, two years of age or older
> wrist and hand disability due to upper limb spasticity associated with stroke in adults
> ankle disability due to lower limb spasticity associated with stroke in adults
• symptomatic relief of blepharospasm, hemifacial spasm and idiopathic cervical dystonia (spasmodic torticollis)
• prophylaxis of headaches in adults with chronic migraine (headaches on at least 15 days per month of which at least 8 days are with migraine)
Bladder disorders:
• management of bladder dysfunctions in adult patients who are not adequately managed with anticholinergics
> overactive bladder with symptoms of urinary incontinence, urgency and frequency
> neurogenic detrusor overactivity with urinary incontinence due to subcervical spinal cord injury (traumatic or non-traumatic), or multiple sclerosis
Skin and skin appendage disorder:
• management of severe hyperhidrosis of the axillae, which does not respond to topical treatment with antiperspirants or antihidrotics
• temporary improvement in the appearance of the following facial lines, when the severity of these lines has an important psychological impact in adult patients:
> moderate to severe vertical lines between the eyebrows seen at maximum frown (glabellar lines)
> moderate to severe lateral canthal lines (crow’s feet lines) seen at maximum smile
> moderate to severe crow’s feet lines seen at maximum smile and glabellar lines seen at maximum frown when treated simultaneously.
4.2 Posology and method of administration
Posology
Botulinum toxin units are not interchangeable from one product to another. Doses recommended in Allergan Units are different from other botulinum toxin preparations.
Elderly patients
Dosages for elderly patients are the same as for younger adults. Initial dosing should begin at the lowest recommended dose for the specific indication. Elderly patients with significant medical history and concomitant medications should be treated with caution.
There is limited data in patients older than 65 years managed with BOTOX for urinary incontinence with neurogenic detrusor overactivity and for facial lines (see section 5.1).
Paediatric population
The safety and efficacy of BOTOX in the treatment of individual indications have not been established in children and adolescents under the ages listed in the table below. No data are available.
|
• Focal spasticity associated with paediatric cerebral palsy |
2 years |
|
• Upper and lower limb spasticity associated with stroke |
18 years |
|
• Blepharospasm/Hemifacial spasm/ Idiopathic Cervical dystonia |
12 years |
|
• Chronic migraine (CM) |
18 years |
|
• Overactive Bladder (OAB) and Neurogenic Detrusor Overactivity (NDO) |
18 years |
|
• Primary hyperhidrosis of the axillae |
12 years (limited experience in adolescents between 12 and 17 years, see sections 4.8 and 5.1) |
|
• Glabellar lines seen at maximum frown and/or crow’s feet lines seen at maximum smile |
18 years |
Method of Administration
BOTOX should only be administered by physicians with appropriate qualifications and expertise in the treatment and the use of the required equipment.
This product is for single use only and any unused solution should be discarded. The most appropriate vial size should be selected for the indication.
An injection volume of approximately 0.1 ml is recommended. A decrease or increase in the BOTOX dose is possible by administering a smaller or larger injection volume. The smaller the injection volume the less discomfort and less spread of toxin in the injected muscle occurs. This is of benefit in reducing effects on nearby muscles when small muscle groups are being injected.
For instructions on reconstitution of the powder for solution for injection, handling and disposal of vials please refer to section 6.6.
Refer to specific guidance for each indication described below.
Generally valid optimum dose levels and number of injection sites per muscle have not been established for all indications. In these cases, individual treatment regimens should therefore be drawn up by the physician. Optimum dose levels should be determined by titration but the recommended maximum dose should not be exceeded.
NEUROLOGIC DISORDERS
Focal spasticity associated with paediatric cerebral palsy
Recommended needle: Sterile 23-26 gauge/0.60-0.45 mm needle.
Administration guidance: To be administered as a divided dose through single
injections into the medial and lateral heads of the
|
Recommended dose: |
affected gastrocnemius muscle. Hemiplegia: the initial recommended total dose is 4 Units/kg body weight in the affected limb. Diplegia: the initial recommended total dose is 6 Units/kg body weight divided between the affected limbs. |
Maximum dose: 200 Units in total
|
Additional information: |
Clinical improvement generally occurs within the first two weeks after injection. Repeat doses should be administered when the clinical effect of a previous injection diminishes but not more frequently than every three months. It may be possible to adapt the dosage regimen to obtain an interval of at least six months between treatment sessions. |
Focal upper limb spasticity associated with stroke
|
Recommended needle: |
Sterile 25, 27 or 30 gauge needle. Needle length should be determined based on muscle location and depth. |
Administration guidance: Localisation of the involved muscles with techniques
such as electromyographic guidance, nerve
|
Recommended dose: |
stimulation, or ultrasound is recommended. Multiple injection sites may allow BOTOX to have more uniform contact with the innervation areas of the muscle and are especially useful in larger muscles. The exact dosage and number of injection sites may be tailored to the individual based on the size, number and location of muscles involved, the severity of spasticity, the presence of local muscle weakness, and the patient response to previous treatment. The following doses are recommended: Muscle Total Dosage; Number of Sites Flexor digitorum profundus 15 - 50 Units; 1-2 sites Flexor digitorum sublimis 15 - 50 Units; 1-2 sites Flexor carpi radialis 15 - 60 Units; 1-2 sites Flexor carpi ulnaris 10 - 50 Units; 1-2 sites Adductor Pollicis 20 Units; 1-2 sites Flexor Pollicis Longus 20 Units; 1-2 sites |
Maximum dose: Between 200 and 240 Units divided among selected
muscles.
Additional information: If it is deemed appropriate by the treating physician,
the patient should be considered for re-injection when the clinical effect of the previous injection has diminished. Re-injections should occur no sooner than 12 weeks after the previous injection. The degree and pattern of muscle spasticity at the time of re-injection may necessitate alterations in the dose of BOTOX and muscles to be injected. The lowest effective dose should be used.
Focal lower limb spasticity associated with stroke
Recommended needle: Sterile 25, 27 or 30 gauge needle. Needle length
should be determined based on muscle location and depth.
Administration guidance: Localisation of the involved muscles with techniques
such as electromyographic guidance, nerve stimulation, or ultrasound is recommended. Multiple injection sites may allow BOTOX to have more uniform contact with the innervation areas of the muscle and are especially useful in larger muscles.
The following diagrams indicate the injection sites for adult lower limb spasticity:
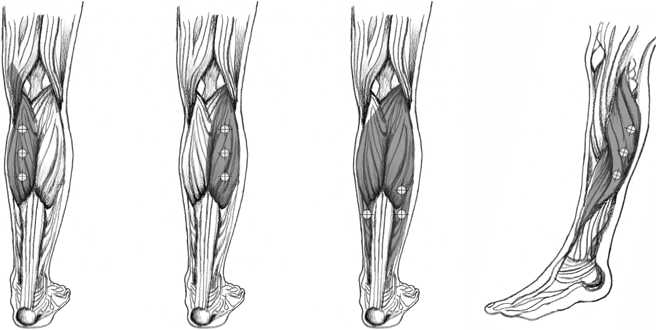
Medial head of Lateral head of Soleus Tibialis posterior
gastrocnemius gastrocnemius
Recommended dose: The recommended dose for treating adult lower limb
spasticity involving the ankle is 300 Units divided among 3 muscles.
|
Muscle |
Recommended Dose Total Dosage; Number of Sites |
|
Gastrocnemius Medial head |
75 Units; 3 sites |
|
Lateral head |
75 Units; 3 sites |
|
Soleus |
75 Units; 3 sites |
|
Tibialis Posterior |
75 Units; 3 sites |
Additional information: If it is deemed appropriate by the treating physician,
the patient should be considered for re-injection when the clinical effect of the previous injection has diminished, but generally no sooner than 12 weeks after the previous injection.
Blepharospasm/hemifacial spasm
Recommended needle: Sterile, 27-30 gauge/0.40-0.30 mm needle.
Administrative guidance: Electromyographic guidance is not necessary.
Recommended dose: The initial recommended dose is 1.25-2.5 Units
(0.05-0.1 ml volume at each site) injected into the medial and lateral orbicularis oculi of the upper lid and the lateral orbicularis oculi of the lower lid. Additional sites in the brow area, the lateral orbicularis and in the upper facial area may also be injected if spasms here interfere with vision.
The following diagrams indicate the possible injection sites:
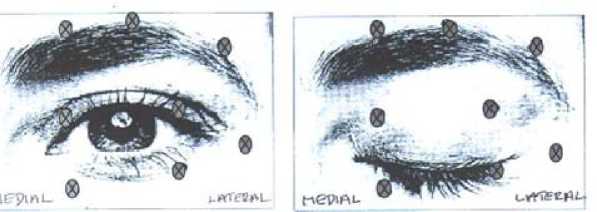
Maximum dose: The initial dose should not exceed 25 Units per eye.
In the management of blepharospasm total dosing should not exceed 100 Units in total every 12 weeks.
Additional information: Avoiding injection near levator palpebrae superioris
may reduce the complication of ptosis. Avoiding medial lower lid injections, and thereby reducing diffusion into the inferior oblique, may reduce the complication of diplopia.
In general, the initial effect of the injections is seen within three days and reaches a peak at one to two weeks post-treatment. Each treatment lasts approximately three months, following which the procedure can be repeated indefinitely. Normally no additional benefit is conferred by treating more frequently than every three months.
At repeat treatment sessions, the dose may be increased up to two-fold if the response from the initial treatment is considered insufficient - usually defined as an effect that does not last longer than two months. However, there appears to be little benefit obtainable from injecting more than 5 Units per site.
Patients with hemifacial spasm or VIIth nerve disorders should be treated as for unilateral blepharospasm, with other affected facial muscles being injected as needed. Electromyographic control may be necessary to identify affected small circumoral muscles.
Cervical dystonia Recommended needle:
Administrative guidance:
Recommended dose:
A 25, 27 or 30 gauge/0.50-0.30 mm needle may be used for superficial muscles, and a 22 gauge needle may be used for deeper musculature.
The treatment of cervical dystonia typically may include injection of BOTOX into the sternocleidomastoid, levator scapulae, scalene, splenius capitis, semispinalis, longissimus and/or the trapezius muscle(s). This list is not exhaustive as any of the muscles responsible for controlling head position may be involved and therefore require treatment. The muscle mass and the degree of hypertrophy are factors to be taken into consideration when selecting the appropriate dose. Muscle activation patterns can change spontaneously in cervical dystonia without a change in the clinical presentation of dystonia.
In case of any difficulty in isolating the individual muscles, injections should be made under electromyographic assistance.
Multiple injection sites allow BOTOX to have more uniform contact with the innervation areas of the dystonic muscle and are especially useful in larger muscles. The optimal number of injection sites is dependent upon the size of the muscle to be chemically denervated.
Dosing must be tailored to the individual patient based on the patient's head and neck position, location of pain, muscle hypertrophy, patient's body weight, and patient response.
Initial dosing in a naive patient should begin at the lowest effective dose.
To minimise the incidence of dysphagia, the stemomastoid should not be injected bilaterally.
The following doses are recommended:
|
Type I Head rotated toward side of shoulder elevation |
Stemomastoid Levator scapulae Scalene Splenius capitis Trapezius |
50 - 100 Units; at least 2 sites 50 Units; 1 - 2 sites 25 - 50 Units; 1 - 2 sites 25 - 75 Units; 1 - 3 sites 25 - 100 Units; 1 - 8 sites |
|
Type II Head rotation only |
Stemomastoid |
25 - 100 Units; at least 2 sites if >25 Units given |
|
Type III Head tilted toward side of shoulder elevation |
Sternomastoid Levator scapulae Scalene Trapezius |
25 - 100 Units at posterior border; at least 2 sites if >25 Units given 25 - 100 Units; at least 2 sites 25 - 75 Units; at least 2 sites 25 - 100 Units; 1 - 8 sites |
|
Type IV Bilateral posterior cervical muscle spasm with elevation of the face |
Splenius capitis and cervicis |
50 - 200 Units; 2 - 8 sites, treat bilaterally (This is the total dose and not the dose for each side of the neck) |
Maximum dose: No more than 50 Units should be given at any one
injection site.
No more than 100 Units should be given to the sternomastoid.
No more than 200 Units in total should be injected for the first course of therapy, with adjustments made in subsequent courses dependent on the initial response, up to a maximum total dose of 300 Units.
Additional information: Treatment intervals of less than 10 weeks are not
recommended.
Chronic migraine
Recommended needle: Sterile 30 gauge, 0.5 inch needle.
A 1 inch needle may be needed in the neck region for patients with extremely thick neck muscles.
Administration guidance: Injections should be divided across 7 specific
head/neck muscle areas as specified in the diagrams below. With the exception of the procerus muscle, which should be injected at 1 site (midline), all muscles should be injected bilaterally with half the number of injection sites administered to the left, and half to the right side of the head and neck.
The following diagrams indicate the injection sites:
2 3 4
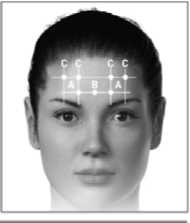
A. Corrugator: 5 U each side
B. Procerus: 5 U (one site)
C. Frontalis: 10 U each side
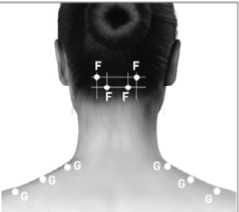
F. Cervical Paraspinal: 10 U each side
D. Temporalis: 20 U each side
E. Occipitalis: 15 U each side
G. Trapezius: 15 U each side
If there is a predominant pain location(s), additional injections to one or both sides may be administered in up to 3 specific muscle groups (occipitalis, temporalis and trapezius), up to the maximum dose per muscle as indicated in the table below.
The following diagrams indicate recommended muscle groups for optional additional injections:
r
D. Temporalis: 5 U/site (<2 additional sites)
E. Occipitalis: 5 U/site (<2 additional sites)
G. Trapezius: 5 U/site («4 additional sites)
Recommended dose:
155 Units to 195 Units administered intramuscularly as 0.1 ml (5 Units) injections to 31 and up to 39 sites.
|
Recommended Dose | |
|
Head/Neck Area |
Total Dosage (number of sites3) |
|
Corrugatorb |
10 Units (2 sites) |
|
Procerus |
5 Units (1 site) |
|
Frontalisb |
20 Units (4 sites) |
|
Temporalisb |
40 Units (8 sites) up to 50 Units (up to 10 sites) |
|
Occipitalisb |
30 Units (6 sites) up to 40 Units (up to 8 sites) |
|
Cervical Paraspinal Muscle Groupb |
20 Units (4 sites) |
|
Trapeziusb |
30 Units (6 sites) up to 50 Units (up to 10 |
|
sites) | |
|
Total Dose Range: |
155 Units to 195 Units 31 to 39 sites |
a1 IM injection site = 0.1 ml = 5 Units BOTOX bDose distributed bilaterally
Additional information: The recommended re-treatment schedule is every 12
weeks.
BLADDER DISORDERS
Overactive bladder
Recommended needle: The injection needle should be filled (primed) with
approximately 1 ml of the reconstituted BOTOX solution prior to the start of the injections (depending on the needle length) to remove any air.
Administration guidance: The reconstituted solution of BOTOX (100 Units/10
ml) is injected via a flexible or rigid cystoscope, avoiding the trigone and base. The bladder should be instilled with enough saline to achieve adequate visualisation for the injections and avoid backflow of the product, but over-distension should be avoided.
The needle should be inserted approximately 2 mm into the detrusor, and 20 injections of 0.5 ml each (total volume 10 ml) should be spaced approximately 1 cm apart (see figure below). For the final injection, approximately 1 ml of 9 mg/ml (0.9%) preservative-free sodium chloride solution for injection should be injected so the full dose is delivered.
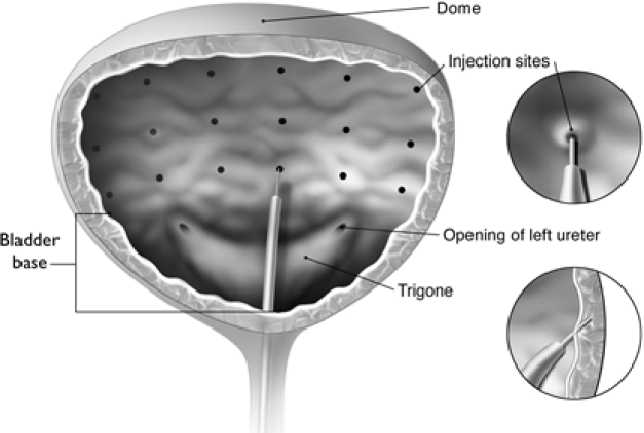
|
Recommended dose: |
The recommended dose is 100 Units of BOTOX, as 0.5 ml (5 Units) injections across 20 sites in the detrusor muscle. |
|
Additional information: |
For the patient preparation and monitoring, see section 4.4. After the injections are given, the saline used for bladder wall visualisation should not be drained so that the patients can demonstrate their ability to void prior to leaving the clinic. The patient should be observed for at least 30 minutes post-injection and until a spontaneous void has occurred. Patients should be considered for reinjection when the clinical effect of the previous injection has diminished but no sooner than 3 months from the prior bladder injection. |
Urinary incontinence due to neurogenic detrusor overactivity
|
Recommended needle: |
The injection needle should be filled (primed) with approximately 1 ml of the reconstituted BOTOX solution prior to the start of the injections (depending on the needle length) to remove any air. |
|
Administration guidance: |
The reconstituted solution of BOTOX (200 Units/30 ml) is injected via a flexible or rigid cystoscope, avoiding the trigone and base. The bladder should be instilled with enough saline to achieve adequate visualisation for the injections and avoid backflow of the product, but over-distension should be avoided. The needle should be inserted approximately 2 mm into the detrusor, and 30 injections of 1 ml each (total volume 30 ml) should be spaced approximately 1 cm apart (see figure above). For the final injection, approximately 1 ml of 9 mg/ml (0.9%) preservative-free sodium chloride solution for injection should be injected so the full dose is delivered. After the injections are given, the saline used for bladder wall visualisation should be drained. |
|
Recommended dose: |
The recommended dose is 200 Units of BOTOX, as 1 ml (~6.7 Units) injections across 30 sites in the detrusor muscle. |
|
Additional information: |
For the patient preparation and monitoring, see section 4.4. Patients should be considered for reinjection when the clinical effect of the previous injection has diminished, but no sooner than 3 months from the prior bladder injection. |
|
Limited data are available beyond 2 treatments. No urodynamic data beyond 2 treatments and no histopathological data after repeated treatment are currently available. Patients should not receive multiple treatments in the event of limited symptomatic improvement. |
SKIN AND SKIN APPENDAGE DISORDERS Primary hyperhidrosis of the axillae
|
Recommended needle: |
Sterile 30 gauge needle. |
|
Administration guidance: |
The hyperhidrotic area to be injected may be defined by using standard staining techniques, e.g. Minor's iodine-starch test. |
|
Recommended dose: |
50 Units of BOTOX is injected intradermally to each axilla, evenly distributed in multiple sites approximately 1-2 cm apart. The recommended injection volume for intradermal injection is 0.1-0.2 ml. |
|
Maximum dose: |
Doses other than 50 Units per axilla cannot be recommended. |
|
Additional information: |
Clinical improvement generally occurs within the first week after injection and persists for 4-7 months. Repeat injection of BOTOX can be administered when the clinical effect of a previous injection diminishes and the treating physician deems it necessary. Injections should not be repeated more frequently than every 16 weeks. |
Glabellar lines seen at maximum frown
|
Recommended needle: |
Sterile 30 gauge needle. |
|
Administration guidance: |
Before injection, the thumb or index finger is to be placed firmly below the orbital rim in order to prevent extravasation below the orbital rim. The needle should be oriented superiorly and medially during the injection. In addition, injections near the levator palpebrae superioris muscle must be avoided, particularly in patients with larger brow-depressor complexes (depressor supercilii). Injections in the corrugator muscle must be done in the central part of that muscle, a distance of at least 1 cm above the arch of the eyebrows (see figure). |
Care should be taken to ensure that BOTOX is not injected into a blood vessel when it is injected in the glabellar lines seen at maximum frown, see section 4.4.
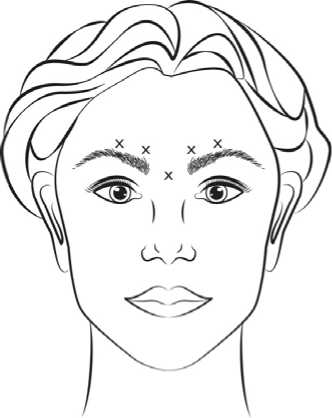
Recommended dose: A volume of 0.1 ml (4 Units) is administered in each
of the 5 injection sites (see Figure): 2 injections in each corrugator muscle and 1 injection in the procerus muscle for a total dose of 20 Units.
Maximum dose: In order to reduce the risk of eyelid ptosis, the
maximum dose of 4 Units for each injection site as well as the number of injection sites should not be exceeded.
Additional Information Treatment intervals should not be more frequent than
every three months. In the event of treatment failure or diminished effect following repeat injections, alternative treatment methods should be employed.
In case of insufficient dose a second treatment session should be initiated by adjusting the total dose up to 40 or 50 Units, taking into account the analysis of the previous treatment failure (see information in All indications).
The efficacy and safety of repeat injections of BOTOX for the treatment of glabellar lines beyond 12 months has not been evaluated.
Crow’s feet lines seen at maximum smile
Recommended needle: Sterile 30 gauge needle.
Administration guidance:
Injections should be given with the needle tip bevel up and oriented away from the eye. The first
injection (A) should be made approximately 1.5 to 2.0 cm temporal to the lateral canthus and just temporal to the orbital rim. If the lines in the crow’s feet region are above and below the lateral canthus, inject as shown in Figure 1. Alternatively, if the lines in the crow’s feet region are primarily below the lateral canthus, inject as shown in Figure 2.
In order to reduce the risk of eyelid ptosis, injections should be made temporal to the orbital rim, thereby maintaining a safe distance from the muscle controlling eyelid elevation.
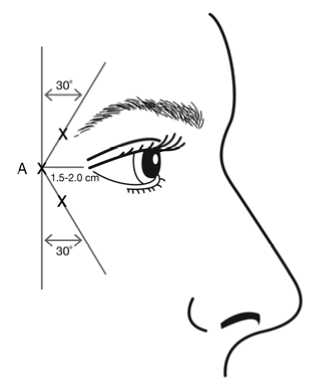
Figure 1: Figure 2:
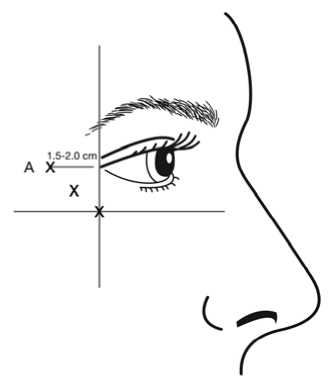
Care should be taken to ensure that BOTOX is not injected into a blood vessel when it is injected in the crow’s feet lines seen at maximum smile (see section 4.4).
Recommended dose: A volume of 0.1 ml (4 Units) is administered in each
of the 3 injection sites per side (total of 6 injection sites) in the lateral orbicularis oculi muscle, for a total dose of 24 Units in a total volume of 0.6 ml (12 Units per side).
For simultaneous treatment with glabellar lines seen at maximum frown, the dose is 24 Units for crow’s feet lines seen at maximum smile and 20 Units for glabellar lines (see Administration guidance for glabellar lines) for a total dose of 44 Units in a total volume of 1.1 ml.
Maximum dose: In order to reduce the risk of eyelid ptosis, the
maximum dose of 4 Units for each injection site as well as the number of injection sites should not be exceeded.
Additional information: Treatment intervals should not be more frequent than
every 3 months.
The efficacy and safety of repeat injections of BOTOX for the treatment of crow’s feet lines beyond 12 months has not been evaluated.
ALL INDICATIONS:
In case of treatment failure after the first treatment session, i.e. absence, at one month after injection, of significant clinical improvement from baseline, the following actions should be taken:
- Clinical verification, which may include electromyographic examination in a specialist setting, of the action of the toxin on the injected muscle(s);
- Analysis of the causes of failure, e.g. bad selection of muscles to be injected, insufficient dose, poor injection technique, appearance of fixed contracture, antagonist muscles too weak, formation of toxin-neutralising antibodies;
- Re-evaluation of the appropriateness of treatment with botulinum toxin type A;
- In the absence of any undesirable effects secondary to the first treatment session, instigate a second treatment session as following: i) adjust the dose, taking into account the analysis of the earlier treatment failure; ii) use EMG; and iii) maintain a three-month interval between the two treatment sessions.
In the event of treatment failure or diminished effect following repeat injections alternative treatment methods should be employed.
4.3 Contraindications
- known hypersensitivity to botulinum toxin type A or to any of the excipients listed in section 6.1;
- presence of infection at the proposed injection site(s).
For the management of bladder disorders:
- urinary tract infection at the time of treatment;
- acute urinary retention at the time of treatment, in patients who are not routinely catheterising;
- patients who are not willing and/or able to initiate catheterisation posttreatment if required;
- presence of bladder calculi.
4.4 Special warnings and precautions for use
The recommended dosages and frequencies of administration of BOTOX should not be exceeded due to the potential for overdose, exaggerated muscle weakness, distant spread of toxin and the formation of neutralising antibodies. Initial dosing in
treatment naive patients should begin with the lowest recommended dose for the specific indication.
Prescribers and patients should be aware that side effects can occur despite previous injections being well tolerated. Caution should therefore be exercised on the occasion of each administration.
Side effects related to spread of toxin distant from the site of administration have been reported (see section 4.8), sometimes resulting in death, which in some cases was associated with dysphagia, pneumonia and/or significant debility.
The symptoms are consistent with the mechanism of action of botulinum toxin and have been reported hours to weeks after injection. The risk of symptoms is probably greatest in patients who have underlying conditions and comorbidities that would predispose them to these symptoms, including children and adults treated for spasticity, and are treated with high doses.
Patients treated with therapeutic doses may also experience exaggerated muscle weakness.
Elderly and debilitated patients should be treated with caution. Generally, clinical studies of BOTOX did not identify differences in responses between the elderly and younger patients except for facial lines (see section 5.1). Dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range.
Consideration should be given to the risk-benefit implications for the individual patient before embarking on treatment with BOTOX.
Dysphagia has also been reported following injection to sites other than the cervical musculature (see section 4.4 ‘Cervical Dystonia’).
BOTOX should only be used with extreme caution and under close supervision in patients with subclinical or clinical evidence of defective neuromuscular transmission e.g. myasthenia gravis or Lambert-Eaton Syndrome in patients with peripheral motor neuropathic diseases (e.g. amyotrophic lateral sclerosis or motor neuropathy) and in patients with underlying neurological disorders. Such patients may have an increased sensitivity to agents such as BOTOX, even at therapeutic doses, which may result in excessive muscle weakness and an increased risk of clinically significant systemic effects including severe dysphagia and respiratory compromise. The botulinum toxin product should be used under specialist supervision in these patients and should only be used if the benefit of treatment is considered to outweigh the risk. Patients with a history of dysphagia and aspiration should be treated with extreme caution.
Patients or caregivers should be advised to seek immediate medical care if swallowing, speech or respiratory disorders arise.
As with any treatment with the potential to allow previously-sedentary patients to resume activities, the sedentary patient should be cautioned to resume activity gradually.
The relevant anatomy, and any alterations to the anatomy due to prior surgical procedures, must be understood prior to administering BOTOX and injection into vulnerable anatomic structures must be avoided.
Pneumothorax associated with injection procedure has been reported following the administration of BOTOX near the thorax.
Caution is warranted when injecting in proximity to the lung (particularly the apices) or other vulnerable anatomic structures.
Serious adverse events including fatal outcomes have been reported in patients who had received off-label injections of BOTOX directly into salivary glands, the oro-lingual-pharyngeal region, oesophagus and stomach. Some patients had pre-existing dysphagia or significant debility.
Serious and/or immediate hypersensitivity reactions have been rarely reported including anaphylaxis, serum sickness, urticaria, soft tissue oedema, and dyspnoea. Some of these reactions have been reported following the use of BOTOX either alone or in conjunction with other products associated with similar reactions. If such a reaction occurs further injection of BOTOX should be discontinued and appropriate medical therapy, such as epinephrine, immediately instituted. One case of anaphylaxis has been reported in which the patient died after being injected with BOTOX inappropriately diluted with 5 ml of 1% lidocaine.
As with any injection, procedure-related injury could occur. An injection could result in localised infection, pain, inflammation, paraesthesia, hypoaesthesia, tenderness, swelling, erythema, and/or bleeding/bruising. Needle-related pain and/or anxiety may result in vasovagal responses, e.g. syncope, hypotension, etc.
Caution should be used when BOTOX is used in the presence of inflammation at the proposed injection site(s) or when excessive weakness or atrophy is present in the target muscle. Caution should also be exercised when BOTOX is used for treatment of patients with peripheral motor neuropathic diseases (e.g., amyotrophic lateral sclerosis or motor neuropathy).
There have been reports of adverse events following administration of BOTOX involving the cardiovascular system, including arrhythmia and myocardial infarction, some with fatal outcomes. Some of these patients had risk factors including preexisting cardiovascular disease.
New onset or recurrent seizures have been reported, typically in patients who are predisposed to experiencing these events. The exact relationship of these events to botulinum toxin injection has not been established. The reports in children were predominantly from cerebral palsy patients treated for spasticity.
Formation of neutralising antibodies to botulinum toxin type A may reduce the effectiveness of BOTOX treatment by inactivating the biological activity of the toxin. Results from some studies suggest that BOTOX injections at more frequent intervals or at higher doses may lead to greater incidence of antibody formation. When appropriate, the potential for antibody formation may be minimised by injecting with the lowest effective dose given at the longest clinically indicated intervals between injections.
Clinical fluctuations during the repeated use of BOTOX (as with all botulinum toxins) may be a result of different vial reconstitution procedures, injection intervals, muscles injected and slightly differing potency values given by the biological test method used.
Paediatric use
The safety and efficacy of BOTOX in indications other than those described for the paediatric population in section 4.1 has not been established. Post-marketing reports of possible distant spread of toxin have been very rarely reported in paediatric
patients with comorbidities, predominantly with cerebral palsy. In general the dose used in these cases was in excess of that recommended (see section 4.8).
There have been rare spontaneous reports of death sometimes associated with aspiration pneumonia in children with severe cerebral palsy after treatment with botulinum toxin, including following off-label use (e.g. neck area). Extreme caution should be exercised when treating paediatric patients who have significant neurologic debility, dysphagia, or have a recent history of aspiration pneumonia or lung disease. Treatment in patients with poor underlying health status should be administered only if the potential benefit to the individual patient is considered to outweigh the risks.
NEUROLOGIC DISORDERS
Focal spasticity associated with _ paediatric cerebral _ palsy and spasticity of the ankle, hand and wrist in adult post-stroke patients
BOTOX is a treatment of focal spasticity that has only been studied in association with usual standard of care regimens, and is not intended as a replacement for these treatment modalities. BOTOX is not likely to be effective in improving range of motion at a joint affected by a fixed contracture.
BOTOX should only be used for the treatment of focal spasticity in adult post-stroke patients if muscle tone reduction is expected to result in improved function (e.g. improvements in gait), or improved symptoms (e.g. reduction in muscle spasms or pain), and/or to facilitate care.
Caution should be exercised when treating adult patients with post-stroke spasticity who may be at increased risk of fall. In clinical studies where patients were treated for lower limb spasticity (some of whom also received concurrent treatment for upper limb spasticity), the incidence of fall was 7.2% and 4.9% of patients in the BOTOX and placebo groups, respectively.
There have been post-marketing reports of death (sometimes associated with aspiration pneumonia) and of possible distant spread of toxin in children with comorbidities, predominantly cerebral palsy following treatment with botulinum toxin. See warnings under section 4.4, ‘Paediatric use’.
Blepharospasm
Reduced blinking following botulinum toxin injection into the orbicularis muscle can lead to corneal exposure, persistent epithelial defect, and corneal ulceration, especially in patients with VII nerve disorders. Careful testing of corneal sensation in eyes previously operated upon, avoidance of injection into the lower lid area to avoid ectropion, and vigorous treatment of any epithelial defect should be employed. This may require protective drops, ointment, therapeutic soft contact lenses, or closure of the eye by patching or other means.
Ecchymosis occurs easily in the soft eyelid tissues. This can be minimised by applying gentle pressure at the injection site immediately after injection.
Because of the anticholinergic activity of botulinum toxin, caution should be exercised when treating patients at risk for angle closure glaucoma, including patients with anatomically narrow angles.
Cervical dystonia
Patients with cervical dystonia should be informed of the possibility of experiencing dysphagia which may be very mild, but could be severe. Dysphagia may persist for two to three weeks after injection, but has been reported to last up to five months post-injection. Consequent to the dysphagia there is the potential for aspiration, dyspnoea and occasionally the need for tube feeding. In rare cases dysphagia followed by aspiration pneumonia and death has been reported.
Limiting the dose injected into the sternocleidomastoid muscle to less than 100 Units may decrease the occurrence of dysphagia. Patients with smaller neck muscle mass, or patients who receive bilateral injections into the sternocleidomastoid muscle, have been reported to be at greater risk of dysphagia. Dysphagia is attributable to the spread of the toxin to the oesophageal musculature. Injections into the levator scapulae may be associated with an increased risk of upper respiratory infection and dysphagia.
Dysphagia may contribute to decreased food and water intake resulting in weight loss and dehydration. Patients with subclinical dysphagia may be at increased risk of experiencing more severe dysphagia following a BOTOX injection.
Chronic migraine
No efficacy has been shown for BOTOX in the prophylaxis of headaches in patients with episodic migraine (headaches on < 15 days per month).
BLADDER DISORDERS
Patient preparation and monitoring
Prophylactic antibiotics should be administered to patients with sterile urine or asymptomatic bacteriuria in accordance with local standard practice.
The decision to discontinue anti-platelet therapy should be subject to local guidance and benefit/risk consideration for the individual patient. Patients on anti-coagulant therapy need to be managed appropriately to decrease the risk of bleeding.
Appropriate medical caution should be exercised when performing the cystoscopy. The patient should be observed for at least 30 minutes post-injection.
In patients who are not regularly practicing catheterisation, post-void residual urine volume should be assessed within 2 weeks post-treatment and periodically as medically appropriate. Patients should be instructed to contact their physician if they experience difficulties in voiding as catheterisation may be required.
Overactive bladder
Prior to injection an intravesical instillation of diluted local anaesthetic, with or without sedation, may be used, per local site practice. If a local anaesthetic instillation is performed, the bladder should be drained and rinsed with sterile saline before the next steps of the injection procedure.
Urinary incontinence due to neurogenic detrusor overactivity BOTOX injection can be performed under general or local anaesthesia with or without sedation. If a local anaesthetic intravesical instillation is performed, the bladder should be drained and rinsed with sterile saline before the next steps of the injection procedure.
Autonomic dysreflexia associated with the procedure can occur and greater vigilance is required in patients known to be at risk.
SKIN AND SKIN APPENDAGE DISORDER
Primary hyperhidrosis of the axillae
Medical history and physical examination, along with specific additional investigations as required, should be performed to exclude potential causes of secondary hyperhidrosis (e.g. hyperthyroidism, phaeochromocytoma). This will avoid symptomatic treatment of hyperhidrosis without the diagnosis and/or treatment of underlying disease.
Glabellar lines seen at maximum _ frown and/or crow’s _ feet lines seen at maximum smile
It is mandatory that BOTOX is used for one single patient treatment only during a single session. The excess of unused product must be disposed of as detailed in section 6.6. Particular precautions should be taken for product preparation and administration as well as for the inactivation and disposal of the remaining unused solution (see section 6.6).
The use of BOTOX is not recommended in individuals under 18 years. There is limited phase 3 clinical data with BOTOX in patients older than 65 years.
Care should be taken to ensure that BOTOX is not injected into a blood vessel when it is injected in the glabellar seen at maximum frown or in the crow’s feet lines seen at maximum smile , see section 4.2. There is a risk of eyelid ptosis following treatment, refer to Section 4.2 for administration instructions on how to minimise this risk.
4.5 Interaction with other medicinal products and other forms of interaction
Theoretically, the effect of botulinum toxin may be potentiated by aminoglycoside antibiotics or spectinomycin, or other medicinal products that interfere with neuromuscular transmission (e.g. neuromuscular blocking agents).
The effect of administering different botulinum neurotoxin serotypes at the same time or within several months of each other is unknown. Excessive neuromuscular weakness may be exacerbated by administration of another botulinum toxin prior to the resolution of the effects of a previously administered botulinum toxin.
No interaction studies have been performed. No interactions of clinical significance have been reported.
There are no data available on the concomitant use of anticholinergics with BOTOX injections in the management of overactive bladder.
4.6 Fertility, pregnancy and lactation Pregnancy
There are no adequate data from the use of botulinum toxin type A in pregnant women. Studies in animals have shown reproductive toxicity (see Section 5.3). The potential risk for humans is unknown. BOTOX is not recommended during pregnancy and in women of childbearing potential not using contraception.
Breast-feeding
There is no information on whether BOTOX is excreted in human milk. The use of BOTOX during breast-feeding cannot be recommended.
Fertility
There are no adequate data on the effects on fertility from the use of botulinum toxin type A in women of childbearing potential. Studies in male and female rats have shown fertility reductions (see section 5.3).
4.7 Effects on ability to drive and use machines
No studies on the effects on the ability to drive and use machines have been performed. However, BOTOX may cause asthenia, muscle weakness, somnolence, dizziness and visual disturbance, which could affect driving and the operation of machinery.
4.8 Undesirable effects
a) General
In controlled clinical trials adverse events considered by the investigators to be related to BOTOX were reported in 35% of the patients with blepharospasm, 28% with cervical dystonia, 17% with paediatric cerebral palsy, 11% with primary hyperhidrosis of the axillae, 16% with focal spasticity of the upper limb associated with stroke, 15% with focal spasticity of the lower limb associated with stroke, 26% with overactive bladder, and 32% with neurogenic detrusor overactivity. In clinical trials for chronic migraine, the incidence was 26% with the first treatment and declined to 11% with a second treatment.
In controlled clinical trials for glabellar lines seen at maximum frown, adverse events considered by the investigators to be related to BOTOX were reported in 23% (placebo 19%) of patients. In treatment cycle 1 of the pivotal controlled clinical trials for crow’s feet lines seen at maximum smile, such events were reported in 8% (24 Units for crow’s feet lines alone) and 6% (44 Units: 24 Units for crow’s feet lines administered simultaneously with 20 Units for glabellar lines) of patients compared to 5% for placebo.
Adverse reactions may be related to treatment, injection technique or both. In general, adverse reactions occur within the first few days following injection and, while generally transient, may have a duration of several months or, in rare cases, longer.
Local muscle weakness represents the expected pharmacological action of botulinum toxin in muscle tissue. However, weakness of adjacent muscles and/or muscles remote from the site of injection has been reported.
As is expected for any injection procedure, localised pain, inflammation, paraesthesia, hypoaesthesia, tenderness, swelling/oedema, erythema, localised infection, bleeding and/or bruising have been associated with the injection. Needle-related pain and/or anxiety have resulted in vasovagal responses, including transient symptomatic hypotension and syncope. Fever and flu syndrome have also been reported after injections of botulinum toxin.
b) Adverse reactions - frequency by indication
The frequency of adverse reactions reported in the clinical trials is defined as follows: Very Common (> 1/10); Common (>1/100 to <1/10); Uncommon (>1/1,000 to <1/100); Rare (>1/10,000 to <1/1,000); Very Rare (<1/10,000).
NEUROLOGIC DISORDERS:
Focal spasticity associated with paediatric cerebral palsy
|
System Organ Class |
Preferred Term |
Frequency |
|
Infections and infestations |
Viral infection, ear infection |
Very Common |
|
Nervous system disorders |
Somnolence, gait disturbance, paraesthesia |
Common |
|
Skin and subcutaneous tissue disorders |
Rash |
Common |
|
Musculoskeletal and connective tissue disorders |
Myalgia, muscular weakness, pain in extremity |
Common |
|
Renal and urinary disorders |
Urinary incontinence |
Common |
|
General disorders and administration site conditions |
Malaise, injection site pain, asthenia |
Common |
|
Injury, poisoning and procedural complications |
Fall |
Common |
Focal upper limb spasticity associated with stroke
|
System Organ Class |
Preferred Term |
Frequency |
|
Psychiatric disorders |
Depression, insomnia |
Uncommon |
|
Nervous system disorders |
Hypertonia |
Common |
|
Hypoasthesia, headache, paraesthesia, incoordination, amnesia |
Uncommon | |
|
Ear and labyrinth disorders |
Vertigo |
Uncommon |
|
Vascular disorders |
Orthostatic hypotension |
Uncommon |
|
Gastrointestinal disorders |
Nausea, oral paraesthesia |
Uncommon |
|
Skin and subcutaneous tissue disorders |
Ecchymosis, purpura |
Common |
|
Dermatitis, pruritus, rash |
Uncommon | |
|
Musculoskeletal and connective tissue disorders |
Pain in extremity, muscle weakness |
Common |
|
Arthralgia, bursitis |
Uncommon | |
|
General disorders and administration site conditions |
Injection site pain, pyrexia, influenza-like illness, injection site haemorrhage, injection site irritation |
Common |
|
Asthenia, pain, injection site hypersensitivity, malaise, peripheral oedema |
Uncommon |
Some of the uncommon events may be disease related.
Focal lower limb spasticity associated with stroke
|
System Organ Class |
Preferred Term |
Frequency |
|
Skin and subcutaneous tissue disorders |
Rash |
Common |
|
Musculoskeletal and connective tissue disorders |
Arthralgia, musculoskeletal stiffness |
Common |
|
General disorders and administration site conditions |
Peripheral oedema |
Common |
Blepharospasm/hemifacial spasm
|
System Organ Class |
Preferred Term |
Frequency |
|
Nervous system disorders |
Dizziness, facial paresis, facial palsy |
Uncommon |
|
Eye disorders |
Eyelid ptosis |
Very Common |
|
Punctate keratitis, lagophthalmos, dry eye, photophobia, eye irritation, lacrimation increase |
Common | |
|
Keratitis, ectropion, diplopia, entropion, visual disturbance, blurred vision |
Uncommon | |
|
Eyelid oedema |
Rare | |
|
Corneal ulceration, corneal epithelium defect, corneal perforation |
Very Rare | |
|
Skin and subcutaneous tissue disorders |
Ecchymosis |
Common |
|
Rash/dermatitis |
Uncommon | |
|
General disorders and administration site conditions |
Irritation, face oedema |
Common |
|
Fatigue |
Uncommon |
Cervical dystonia
|
System Organ Class |
Preferred Term |
Frequency |
|
Infections and infestations |
Rhinitis, upper respiratory infection |
Common |
|
Nervous system disorders |
Dizziness, hypertonia, hypoaesthesia, somnolence, headache |
Common |
|
Eye disorders |
Diplopia, eyelid ptosis |
Uncommon |
|
Respiratory, thoracic and mediastinal disorders |
Dyspnoea, dysphonia |
Uncommon |
|
Gastrointestinal disorders |
Dysphagia |
Very common |
|
Dry mouth, nausea |
Common | |
|
Musculoskeletal and connective tissue disorders |
Muscular weakness |
Very common |
|
Musculoskeletal stiffness and musculoskeletal soreness |
Common | |
|
General disorders and administration site conditions |
Pain |
Very common |
|
Asthenia, influenza-like illness, malaise |
Common | |
|
Pyrexia |
Uncommon |
Chronic migraine
|
System Organ Class |
Preferred Term |
Frequency |
|
Nervous system disorders |
Headache*, migraine*, facial paresis |
Common |
|
Eye disorders |
Eyelid ptosis |
Common |
|
Eyelid oedema |
Uncommon | |
|
Gastrointestinal disorders |
Dysphagia |
Uncommon |
|
Skin and subcutaneous tissue disorders |
Pruritis, rash |
Common |
|
Pain of skin |
Uncommon | |
|
Musculoskeletal and connective tissue disorders |
Neck pain, myalgia, musculoskeletal pain, musculoskeletal stiffness, muscle spasms, muscle tightness, muscular weakness |
Common |
|
Pain in jaw |
Uncommon | |
|
General disorders and administration site conditions |
Injection site pain |
Common |
* In placebo-controlled trials, headache and migraine, including serious cases of intractable or worsening of headache/migraine, were reported more frequently with BOTOX (9%) than with placebo (6%). They typically occurred within the first month after the injections and their incidence declined with repeated treatments.
BLADDER DISORDERS:
Overactive bladder
|
System Organ Class |
Preferred Term |
Frequency |
|
Infections and infestations |
Urinary tract infection |
Very common |
|
Bacteriuria |
Common | |
|
Renal and urinary disorders |
Dysuriaf |
Very common |
|
Urinary retention, residual urine volume*, pollakiuria, leukocyturia |
Common |
*elevatedpost-void residual urine volume (PVR) not requiring catheterisation fprocedure-related adverse reactions
In the phase 3 clinical trials urinary tract infection was reported in 25.5% of patients treated with BOTOX 100 Units and 9.6% of patients treated with placebo. Urinary retention was reported in 5.8% of patients treated with BOTOX 100 Units and in 0.4% of patients treated with placebo. Clean intermittent catheterisation was initiated in 6.5% of patients following treatment with BOTOX 100 Units versus 0.4% in the placebo group.
Overall, 42.5% of patients (n = 470) were > 65 years of age and 15.1% (n = 167) were > 75 years of age. No overall difference in the safety profile following BOTOX treatment was observed between patients > 65 years compared to patients < 65 years in these studies, with the exception of urinary tract infection where the incidence was higher in elderly patients in both the placebo and BOTOX groups compared to the younger patients.
No change was observed in the overall safety profile with repeat dosing.
Urinary incontinence due to neurogenic detrusor overactivity
|
System Organ Class |
Preferred Term |
Frequency |
|
Infections and infestations |
Urinary tract infection |
Very Common |
|
Psychiatric disorders |
Insomniaf |
Common |
|
Gastrointestinal disorders |
Constipationf |
Common |
|
Musculoskeletal and connective tissue disorders |
Muscular weaknessf, muscle spasm |
Common |
|
Renal and urinary disorders |
Urinary retention |
Very Common |
|
Haematuria*, bladder diverticulum |
Common | |
|
General disorders and administration site conditions |
Fatiguef, gait disturbancef |
Common |
|
Injury, poisoning and procedural complications |
Autonomic dysreflexia*, fallf |
Common |
* procedure-related adverse reactions f only in multiple sclerosis
In the phase 3 clinical trials, urinary tract infection was reported in 49% of patients treated with BOTOX 200 Units and in 36% of patients treated with placebo (in multiple sclerosis patients: 53% vs. 29%, respectively; in spinal cord injury patients: 45% vs. 42%, respectively). Urinary retention was reported in 17% of patients treated with BOTOX 200 Units and in 3% of patients treated with placebo (in multiple sclerosis patients: 29% vs. 4%, respectively; in spinal cord injury patients: 5% vs. 1%, respectively). Among patients who were not catheterising at baseline prior to treatment, catheterisation was initiated in 39% following treatment with BOTOX 200 Units versus 17% on placebo. The risk of urinary retention increased in patients older than 65 years.
No change in the type and frequency of adverse reactions was observed following 2 treatments.
SKIN AND SKIN APPENDAGE DISORDER:
Primary hyperhidrosis of the axillae
|
System Organ Class |
Preferred Term |
Frequency |
|
Nervous system disorders |
Headache, paraesthesia |
Common |
|
Vascular disorders |
Hot flushes |
Common |
|
Gastrointestinal disorders |
Nausea |
Uncommon |
|
Skin and subcutaneous tissue disorders |
Hyperhidrosis (non axillary sweating), abnormal skin odour, pruritus, subcutaneous nodule, alopecia |
Common |
|
Musculoskeletal and connective tissue disorders |
Pain in extremity |
Common |
|
Muscular weakness, myalgia, arthropathy |
Uncommon | |
|
General disorders and administration site conditions |
Injection site pain |
Very common |
|
Pain, injection site oedema, injection site haemorrhage, injection site hypersensitivity, injection site irritation, asthenia, injection site reactions |
Common |
Increase in non axillary sweating was reported in 4.5% of patients within 1 month after injection and showed no pattern with respect to anatomical sites affected. Resolution was seen in approximately 30% of the patients within four months.
Weakness of the arm has been also reported uncommonly (0.7%) and was mild, transient, did not require treatment and recovered without sequelae. This adverse event may be related to treatment, injection technique, or both. In the uncommon event of muscle weakness being reported a neurological examination may be considered. In addition, a re-evaluation of injection technique prior to subsequent injection is advisable to ensure intradermal placement of injections.
In an uncontrolled safety study of BOTOX (50 Units per axilla) in paediatric patients 12 to 17 years of age (n= 144), adverse reactions occurring in more than a single patient (2 patients each) comprised injection site pain and hyperhidrosis (non-axillary sweating).
Glabellar lines
|
System Organ Class |
Preferred Term |
Frequency |
|
Infections and infestations |
Infection |
Uncommon |
|
Psychiatric disorders |
Anxiety |
Uncommon |
|
Nervous system disorders |
Headache |
Common |
|
Paraesthesia, dizziness |
Uncommon | |
|
Eye disorders |
Eyelid ptosis |
Common |
|
Blepharitis, eye pain, visual disturbance |
Uncommon | |
|
Gastrointestinal disorders |
Nausea, oral dryness |
Uncommon |
|
Skin and subcutaneous tissue disorders |
Erythema |
Common |
|
Skin tightness, oedema (face, eyelid, periorbital), photosensitivity reaction, pruritus, dry skin |
Uncommon | |
|
Musculoskeletal and connective tissue disorders |
Localised muscle weakness |
Common |
|
Muscle twitching |
Uncommon | |
|
General disorders and administration site conditions |
Face pain |
Common |
|
Flu syndrome, asthenia, fever |
Uncommon |
Crow’s feet lines
The following adverse drug reactions were reported in the double-blind, placebo-controlled clinical studies following injection of BOTOX 24 Units for crow’s feet lines alone:
|
System Organ Class |
Preferred Term |
Frequency |
|
Eye disorders |
Eyelid oedema |
Common |
|
General disorders and administration site conditions |
Injection site haemorrhage*, injection site haematoma* |
Common |
|
Injection site pain*, injection site paraesthesia |
Uncommon |
*procedure-related adverse reactions
Crow’s feet lines and glabellar lines
The following adverse drug reactions were reported in double-blind, placebo-controlled clinical studies following injection of BOTOX 44 Units (simultaneous treatment of crow’s feet lines and glabellar lines):
|
System Organ Class |
Preferred Term |
Frequency |
|
General disorders and administration site conditions |
Injection site haematoma* |
Common |
|
Injection site haemorrhage*, injection site pain* |
Uncommon |
*procedure-related adverse reactions
No change was observed in the overall safety profile following repeat dosing.
c) Additional information
The following list includes adverse drug reactions or other medically relevant adverse events that have been reported since the drug has been marketed, regardless of indication, and may be in addition to those cited in section 4.4 (Special warnings and precautions for use), and section 4.8 (Undesirable effects).
|
System Organ Class |
Preferred Term |
|
Immune system disorders |
Anaphylaxis, angioedema, serum sickness, urticaria |
|
Metabolism and nutrition disorders |
Anorexia |
|
Nervous system disorders |
Brachial plexopathy, dysphonia, dysarthria, facial paresis, hypoaesthesia, muscle weakness, myasthenia gravis, peripheral neuropathy, paraesthesia, radiculopathy, seizures, syncope, facial palsy |
|
Eye disorders |
Angle-closure glaucoma (for treatment of blepharospasm), lagophthalmos, strabismus, blurred vision, visual disturbance |
|
Ear and labyrinth disorders |
Hypoacusis, tinnitus, vertigo |
|
Cardiac disorders |
Arrhythmia, myocardial infarction |
|
Respiratory, thoracic and mediastinal disorders |
Aspiration pneumonia (some with fatal outcome), dyspnoea, respiratory depression, respiratory failure |
|
Gastrointestinal disorders |
Abdominal pain, diarrhoea, constipation, dry mouth, dysphagia, nausea, vomiting |
|
Skin and subcutaneous tissue disorders |
Alopecia, dermatitis psoriasiform, erythema multiforme, hyperhidrosis, madarosis, pruritus, rash |
|
Musculoskeletal and connective tissue disorders |
Muscle atrophy, myalgia |
|
General disorders and administration site |
Denervation atrophy, malaise, pyrexia |
conditions
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Yellow Card Scheme, Website: www.mhra.gov.uk/yellowcard.
4.9 Overdose
Overdose of BOTOX is a relative term and depends upon dose, site of injection, and underlying tissue properties. No cases of systemic toxicity resulting from accidental injection of BOTOX have been observed. Excessive doses may produce local, or distant, generalised and profound neuromuscular paralysis.
No cases of ingestion of BOTOX have been reported.
Signs and symptoms of overdose are not apparent immediately post-injection. Should accidental injection or ingestion occur or overdose be suspected, the patient should be medically monitored for up to several weeks for progressive signs and symptoms of muscular weakness, which could be local or distant from the site of injection and may include ptosis, diplopia, dysphagia, dysarthria, generalised weakness or respiratory failure. These patients should be considered for further medical evaluation and appropriate medical therapy immediately instituted, which may include hospitalisation.
If the musculature of the oropharynx and oesophagus are affected, aspiration may occur which may lead to development of aspiration pneumonia. If the respiratory muscles become paralysed or sufficiently weakened, intubation and assisted respiration will be required until recovery takes place and may involve the need for a tracheostomy and prolonged mechanical ventilation, in addition to other general supportive care.
5 PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic propertie s
ATC class M03A X01 and ATC class D11AX.
The active constituent in BOTOX is a protein complex derived from Clostridium botulinum. The protein consists of type A neurotoxin and several other proteins. Under physiological conditions it is presumed that the complex dissociates and releases the pure neurotoxin.
Clostridium botulinum toxin type A neurotoxin complex blocks peripheral acetyl choline release at presynaptic cholinergic nerve terminals.
Intramuscular injection of the neurotoxin complex blocks cholinergic transport at the neuromuscular junction by preventing the release of acetylcholine. The nerve endings of the neuromuscular junction no longer respond to nerve impulses and secretion of the chemotransmitter is prevented (chemical denervation). Reestablishment of impulse transmission is by newly formed nerve endings and motor end plates. Recovery after intramuscular injection takes place normally within 12 weeks of injection as nerve terminals sprout and reconnect with the endplates.
After intradermal injection, where the target is the eccrine sweat glands, the effect lasted for about 4-7 months in patients treated with 50 Units per axilla.
There is limited clinical trial experience of the use of BOTOX in primary axillary hyperhidrosis in adolescents between the ages of 12 and 18. A single, year long, uncontrolled, repeat dose, safety study was conducted in US paediatric patients 12 to 17 years of age (N=144) with severe primary hyperhidrosis of the axillae.
Participants were primarily female (86.1%) and Caucasian (82.6%). Participants were treated with a dose of 50 Units per axilla for a total dose of 100 Units per patient per treatment. However, no dose finding studies have been conducted in adolescents so no recommendation on posology can be made. Efficacy and safety of BOTOX in this group have not been established.
BOTOX blocks the release of neurotransmitters associated with the genesis of pain. The presumed mechanism for headache prophylaxis is by blocking peripheral signals to the central nervous system, which inhibits central sensitisation, as suggested by pre-clinical and clinical pharmacodynamic studies.
Following intradetrusor injection, BOTOX affects the efferent pathways of detrusor activity via inhibition of acetylcholine release. In addition BOTOX inhibits afferent neurotransmitters and sensory pathways.
Clinical efficacy and safety
NEUROLOGIC DISORDERS
Focal upper limb spasticity associated with stroke
In controlled and open, non-controlled studies, doses between 200 and 240 Units in wrist and flexor muscles were divided among the selected muscles at a given treatment session. In controlled studies, improvement in muscle tone occurred within two weeks with the peak effect generally seen within four to six weeks. In an open, non-controlled continuation study, most patients were re-injected after an interval of 12 to 16 weeks, when the effect on muscle tone had diminished. These patients received up to four injections with a maximal cumulative dose of 960 Units over 54 weeks.
Focal lower limb spasticity associated with stroke
A double-blind, placebo-controlled, randomised, multi-centre, phase 3 clinical study was conducted in adult post-stroke patients with lower limb spasticity affecting the ankle. A total of 120 patients were randomised to receive either BOTOX (n=58; total dose of 300 Units) or placebo (n=62).
Significant improvement compared to placebo was observed in the primary endpoint for the overall change from baseline up to week 12 in Modified Ashworth Scale (MAS) ankle score, which was calculated using the area under the curve (AUC) approach. Significant improvements compared to placebo were also observed for the
mean change from baseline in MAS ankle score at individual post-treatment visits at weeks 4, 6 and 8. The proportion of responders (patients with at least a 1-grade improvement) was also significantly higher (67%-68%) than in placebo-treated patients (31%-36%) at these visits.
BOTOX treatment was also associated with significant improvement in the investigator’s clinical global impression (CGI) of functional disability compared to placebo although the difference was not significant for the patient’s CGI.
Cervical dystonia
In initial controlled clinical trials to establish safety and efficacy for cervical dystonia, doses of reconstituted BOTOX ranged from 140 to 280 Units. In more recent studies, doses ranged from 95 to 360 Units (with an approximate mean of 240 Units).
Clinical improvement generally occurs within the first two weeks after injection. The maximum clinical benefit generally occurs by six weeks post-injection. The duration of beneficial effect reported in clinical studies showed substantial variation (from 2 to 33 weeks) with a typical duration of approximately 12 weeks.
Chronic migraine
Chronic migraine patients without any concurrent headache prophylaxis who, during a 28-day baseline, had at least 4 episodes and > 15 headache days (with at least 4 hours of continuous headache) with at least 50% being migraine/probable migraine, were studied in two Phase 3 clinical trials. Patients were allowed to use acute headache treatments and 66% overused acute treatments during the baseline period.
During the double-blind phase of the trials, the main results achieved after two BOTOX treatments administered at a 12-week interval are shown in the table below.
|
Mean change from baseline at Week 24 |
BOTOX N=688 |
Placebo N=696 |
P-value |
|
Frequency of headache days |
-8.4 |
-6.6 |
< 0.001 |
|
Frequency of moderate/severe headache days |
-7.7 |
-5.8 |
< 0.001 |
|
Frequency of migraine/probable migraine days |
-8.2 |
-6.2 |
< 0.001 |
|
% patients with 50% reduction in headache days |
47% |
35% |
< 0.001 |
|
Total cumulative hours of headache on headache days |
120 |
80 |
< 0.001 |
|
Frequency of headache episodes |
-5.2 |
-4.9 |
0.009 |
|
Total HIT-6* scores |
-4.8 |
-2.4 |
< 0.001 |
* Headache Impact Test
The treatment effect appeared smaller in the subgroup of male patients (n=188) than in the whole study population.
BLADDER DISORDERS
Overactive bladder
Two double-blind, placebo-controlled, randomised, 24-week phase 3 clinical studies were conducted in patients with overactive bladder with symptoms of urge urinary incontinence, urgency, and frequency. A total of 1105 patients (mean age of 60 years), whose symptoms had not been adequately managed with at least one anticholinergic therapy (inadequate response or intolerable side effects), were randomised to receive either 100 Units of BOTOX (n=557), or placebo (n=548), after having discontinued anticholinergics for more than one week.
Primary and Secondary Endpoints at Baseline and Change from Baseline in Pooled Pivotal Studies:
|
Botox |
Placebo |
P-value | |
|
100 Units (N=557) |
(N=548) | ||
|
Daily Frequency of Urinary Incontinence Episodes Mean Baseline |
5.49 |
5.39 | |
|
Mean Change^ at Week 2 |
-2.66 |
-1.05 |
< 0.001 |
|
Mean Change^ at Week 6 |
-2.97 |
-1.13 |
< 0.001 |
|
Mean Change^ at Week 12a |
-2.74 |
-0.95 |
< 0.001 |
|
Proportion with Positive Treatment Response using Treatment Benefit Scale (%) Week 2 |
64.4 |
34.7 |
< 0.001 |
|
Week 6 |
68.1 |
32.8 |
< 0.001 |
|
Week 12a |
61.8 |
28.0 |
<0.001 |
|
Daily Frequency of Micturition Episodes Mean Baseline |
11.99 |
11.48 | |
|
Mean Change^ at Week 12b |
-2.19 |
-0.82 |
< 0.001 |
|
Daily Frequency of Urgency Episodes Mean Baseline |
8.82 |
8.31 | |
|
Mean Change^ at Week 12b |
-3.08 |
-1.12 |
< 0.001 |
|
Incontinence Quality of Life Total Score Mean Baseline |
34.1 |
34.7 | |
|
Mean Change^ at Week 12bc |
+21.3 |
+5.4 |
< 0.001 |
|
King’s Health Questionnaire: Role Limitation Mean Baseline |
65.4 |
61.2 | |
|
Mean Change^ at Week 12bc |
-24.3 |
-3.9 |
< 0.001 |
|
King’s Health Questionnaire: Social Limitation Mean Baseline |
44.8 |
42.4 | |
|
Mean Change^ at Week 12bc |
-16.1 |
-2.5 |
< 0.001 |
|
Percentage of patients achieving full continence at Week 12 (dry patients over a 3-day diary) |
27.1% |
8.4% |
< 0.001 |
|
Percentage of patients achieving reduction from baseline in urinary incontinence episodes at Week 12 | |||
|
at least 75% |
46.0% |
17.7% | |
|
at least 50% |
60.5% |
31.0% |
T Least Squares (LS) mean changes are presented
a Co-primary endpoints b Secondary endpoints
c Pre-defined minimally important change from baseline was +10 points for I-QOL and -5 points for KHQ
The median duration of response following BOTOX treatment, based on patient request for re-treatment, was 166 days (~24 weeks).
A total of 839 patients were evaluated in a long-term open-label extension study. For all efficacy endpoints, patients experienced consistent response with re-treatments. The mean reductions from baseline in daily frequency of urinary incontinence were -3.07 (n=341), -3.49 (n=292), and -3.49 (n=204) episodes at week 12 after the first, second, and third BOTOX 100 Unit treatments, respectively. The corresponding proportions of patients with a positive treatment response on the Treatment Benefit Scale were 63.6% (n=346), 76.9% (n=295), and 77.3% (n=207), respectively.
In the pivotal studies, none of the 615 patients with analysed serum specimens developed neutralising antibodies after 1 - 3 treatments.
Urinary incontinence due to neurogenic detrusor overactivity
Two double-blind, placebo-controlled, randomised phase 3 clinical studies were conducted in a total of 691 patients with spinal cord injury or multiple sclerosis, who were not adequately managed with at least one anticholinergic agent and were either spontaneously voiding or using catheterisation. These patients were randomised to receive either 200 Units of BOTOX (n=227), 300 Units of BOTOX (n=223), or placebo (n=241).
Primary and Secondary Endpoints at Baseline and Change from Baseline in Pooled Pivotal Studies:
|
BOTOX 200 Units |
Placebo |
P-value | |
|
(N=227) |
(N=241) | ||
|
Weekly Frequency of Urinary Incontinence Mean Baseline |
32.4 |
31.5 | |
|
Mean Change^ at Week 2 |
-16.8 |
-9.1 |
<0.001 |
|
Mean Change^ at Week 6a |
-20.0 |
-10.5 |
<0.001 |
|
Mean Change^ at Week 12 |
-19.8 |
-9.3 |
<0.001 |
|
Maximum Cystometric Capacity (ml) | |||
|
Mean Baseline |
250.2 |
253.5 | |
|
Mean Change^ at Week 6b |
+140.4 |
+6.9 |
<0.001 |
|
Maximum Detrusor Pressure during 1st Involuntary Detrusor Contraction (cmH20) | |||
|
Mean Baseline |
51.5 |
47.3 | |
|
Mean Change^ at Week 6b |
-27.1 |
-0.4 |
<0.001 |
|
Incontinence Quality of Life Total Scorec,d | |||
|
Mean Baseline |
35.4 |
35.3 | |
|
Mean Change^ at Week 6b |
+23.6 |
+8.9 |
<0.001 |
|
Mean Change^ at Week 12 |
+26.9 |
+7.1 |
<0.001 |
|
Percentage of patients achieving full continence at Week 6 |
37% |
9% | |
|
(dry patients over a 7 day diary) | |||
|
Percentage of patients achieving reduction from baseline in urinary incontinence episodes at Week 6 | |||
|
at least 75% |
63% |
24% | |
|
at least 50% |
76% |
39% |
T LS mean changes are presented a Primary endpoint b Secondary endpoints
c I-QOL total score scale ranges from 0 (maximum problem) to 100 (no problem at all).
d In the pivotal studies, the pre-specified minimally important difference (MID) for I-QOL total score was 8 points based on MID estimates of 4-11 points reported in neurogenic detrusor overactivity patients.
The median duration of response (time to < 50% reduction in incontinence episodes) was 42 weeks in the 200 Unit dose group. The median interval between the first and second administrations was 42 weeks in patients with spinal cord injury and 45 weeks in patients with multiple sclerosis.
For all efficacy endpoints in the pivotal phase 3 studies, patients experienced consistent response with re-treatment (n=116).
None of the 475 patients with analysed serum specimens developed neutralising antibodies after 1-2 treatments.
In the multiple sclerosis (MS) patients enrolled in the pivotal studies, the MS exacerbation annualised rate (i.e., number of MS exacerbation events per patient year) was 0.23 in the 200 Unit dose group and 0.20 in the placebo group. With repeated BOTOX treatments, including data from a long term study, the MS exacerbation annualised rate was 0.19 during each of the first two BOTOX treatment cycles.
SKIN AND SKIN APPENDAGE DISORDER
Glabellar lines
537 patients with moderate to severe glabellar lines between the eyebrows seen at maximum frown have been included in clinical studies.
BOTOX injections significantly reduced the severity of glabellar lines seen at maximum frown for up to 4 months, as measured by the investigator assessment of glabellar line severity at maximum frown and by subject’s global assessment of change in appearance of his/her glabellar lines seen at maximum frown.
Improvement generally occurred within one week of treatment. None of the clinical endpoints included an objective evaluation of the psychological impact. Thirty days after injection, 80% (325/405) of BOTOX-treated patients were considered by investigators as treatment responders (none or mild severity at maximum frown), compared to 3% (4/132) of placebo-treated patients. At this same timepoint, 89% (362/405) of BOTOX-treated patients felt they had a moderate or better improvement, compared to 7% (9/132) of placebo-treated patients.
BOTOX injections also significantly reduced the severity of glabellar lines at rest. Of the 537 patients enrolled, 39% (210/537) had moderate to severe glabellar lines at rest (15% had no lines at rest). Of these, 74% (119/161) of BOTOX-treated patients were considered treatment responders (none or mild severity) thirty days after injection, compared with 20% (10/49) of placebo-treated patients.
There is limited phase 3 clinical data with BOTOX in patients older than 65 years. Only 6.0% (32/537) of subjects were >65 years old and efficacy results obtained were lower in this population.
Crow’s feet lines
1362 patients with moderate to severe crow’s feet lines seen at maximum smile, either alone (n=445, Study 191622-098) or also with moderate to severe glabellar lines seen at maximum frown (n=917, Study 191622-099), were enrolled.
BOTOX injections significantly reduced the severity of crow’s feet lines seen at maximum smile compared to placebo at all timepoints (p <0.001) for up to 5 months (median 4 months). Improvement assessed by the investigator occurred within one week of treatment. This was measured by the proportion of patients achieving a crow’s feet lines severity rating of none or mild at maximum smile in both pivotal studies; until day 150 (end of study) in Study 191622-098 and day 120 (end of first treatment cycle) in Study 191622-099. For both investigator and subject assessments, the proportion of subjects achieving none or mild crow’s feet lines severity seen at maximum smile was greater in patients with moderate crow’s feet lines seen at maximum smile at baseline, compared to patients with severe crow’s feet lines seen at maximum smile at baseline. Table 1 summarises results at day 30, the timepoint of the primary efficacy endpoint.
In Study 191622-104 (extension to Study 191622-099), 101 patients previously randomised to placebo were enrolled to receive their first treatment at the 44 Units dose. Patients treated with BOTOX had a statistically significant benefit in the primary efficacy endpoint compared to placebo at day 30 following their first active treatment. The response rate was similar to the 44 Units group at day 30 following first treatment in Study 191622-099. A total of 123 patients received 4 cycles of 44 Units BOTOX for combined crow’s feet and glabellar lines treatment.
Day 30: Investigator and Patient Assessment of Crow’s Feet Lines Seen at Maximum Smile - Responder Rates (% of Patients Achieving Crow’s Feet Lines Severity Rating of None or Mild)_
|
Clinical Study |
Dose |
BOTOX |
Placebo |
BOTOX |
Placebo |
|
Investigator Assessment |
Patient Assessment | ||||
|
191622 098 |
24 Units (crow’s feet lines) |
66.7%* (148/222) |
6.7% (15/223) |
58.1%* (129/222) |
5.4% (12/223) |
|
191622 099 |
24 Units (crow’s feet lines) |
54.9%* (168/306) |
3.3% (10/306) |
45.8%* (140/306) |
3.3% (10/306) |
|
44 Units (24 Units crow’s feet lines; 20 Units glabellar lines) |
59.0%* (180/305) |
3.3% (10/306) |
48.5%* (148/305) |
3.3% (10/306) | |
*p < 0.001 (BOTOX vs placebo)
Improvements from baseline in subject-assessment of the appearance of crow’s feet lines seen at maximum smile were seen for BOTOX (24 Units and 44 Units) compared to placebo, at day 30 and at all timepoints following each treatment cycle in both pivotal studies (p<0.001).
Treatment with BOTOX 24 Units also significantly reduced the severity of crow’s feet lines at rest. Of the 528 patients treated, 63% (330/528) had moderate to severe crow’s feet lines at rest at baseline. Of these, 58% (192/330) of BOTOX-treated patients were considered treatment responders (none or mild severity) thirty days after injection, compared with 11% (39/352) of placebo-treated patients.
Improvements in subject’s self-assessment of age and attractiveness were also seen for BOTOX (24 Units and 44 Units) compared to placebo using the Facial Line Outcomes (FLO-11) questionnaire, at the primary timepoint of day 30 (p<0.001) and at all subsequent timepoints in both pivotal studies.
In the pivotal studies, 3.9% (53/1362) of patients were older than 65 years of age. Patients in this age group had a treatment response as assessed by the investigator, of 36% (at day 30) for BOTOX (24 Units and 44 Units).When analysed by age groups of <50 years and >50 years, both populations demonstrated statistically significant improvements compared to placebo. Treatment response for BOTOX 24 Units, as assessed by the investigator, was lower in the group of subjects >50 years of age than those <50 years of age (42.0% and 71.2%, respectively).
Overall BOTOX treatment response for crow’s feet lines seen at maximum smile is lower (60%) than that observed with treatment for glabellar lines seen at maximum frown (80%).
916 patients (517 patients at 24 Units and 399 patients at 44 Units) treated with BOTOX had specimens analysed for antibody formation. No patients developed the presence of neutralising antibodies.
5.2 Pharmacokinetic properties
a) General characteristics of the active substance:
Classical absorption, distribution, biotransformation and elimination studies on the active substance have not been performed due to the extreme toxicity of botulinum toxin type A.
b) Characteristics in patients:
Human ADME studies have not been performed due to the nature of the product. It is believed that little systemic distribution of therapeutic doses of BOTOX occurs. BOTOX is probably metabolised by proteases and the molecular components recycled through normal metabolic pathways.
5.3 Preclinical safety data
Non-clinical data based on conventional studies of safety pharmacology, repeated dose toxicity and genotoxicity reveal no special hazard for humans other than exaggerated pharmacological effects predictable at high doses, given the neurotoxic nature of BOTOX. Carcinogenicity studies have not been conducted.
Acute toxicity
In monkeys receiving a single intramuscular (i.m.) injection of BOTOX, the No Observed Effect Level (NOEL) ranged from 4 to 24 Units/kg. The i.m. LD50 was reported to be 39 Units/kg.
Toxicity on repeated injection
In three different studies (six months in rats; 20 weeks in juvenile monkeys; 1 year in monkeys) where the animals received i.m. injections, the NOEL was at the following respective BOTOX dosage levels: < 4 Units/kg, 8 Units/kg and 4 Units/kg. The main systemic effect was a transient decrease in body weight gain.
There was no indication of a cumulative effect in the animal studies when BOTOX was given at dosage intervals of 1 month or greater.
Decrease in bodyweight was observed following a single intradetrusor injection of <10 Units/kg BOTOX in rats. To simulate inadvertent injection, a single dose of BOTOX (~7 Units/kg) was administered into the prostatic urethra and proximal rectum, the seminal vesicle and urinary bladder wall, or the uterus of monkeys (~3 Units/kg) without adverse clinical effects. However, bladder stones have been observed in monkeys given a single dose of BOTOX to the prostatic urethra and proximal rectum, and in a repeated dose intraprostatic study. Due to anatomical differences the clinical relevance of these findings is unknown. In a 9 month repeat dose intradetrusor study (4 injections), eyelid ptosis was observed at 24 Units/kg, and mortality was observed at doses >24 Units/kg. No adverse effects were observed in monkeys at 12 Units/kg, which corresponds to a 3-fold greater exposure than the recommended clinical dose of 200 Units for urinary incontinence due to neurogenic detrusor overactivity (based on a 50 kg person).
Local toxicity
BOTOX was shown not to cause ocular or dermal irritation, or give rise to toxicity when injected into the vitreous body in rabbits.
Allergic or inflammatory reactions in the area of the injection sites are rarely observed after BOTOX administration. However, formation of haematoma may occur.
Reproduction toxicology
Teratogenic effects
When pregnant mice and rats were injected intramuscularly during the period of organogenesis, the developmental NOEL of BOTOX was at 4 Units/kg. Reductions in ossification were observed at 8 and 16 Units/kg (mice) and reduced ossification of the hyoid bone at 16 Units/kg (rats). Reduced foetal body weights were observed at 8 and 16 Units/kg (rats).
In a range-finding study in rabbits, daily injections at dosages of 0.5 Units/kg/day (days 6 to 18 of gestation), and 4 and 6 Units/kg (administered on days 6 and 13 of gestation), caused death and abortions among surviving dams. External malformations were observed in one foetus each in the 0.125 Units/kg/day and the 2 Units/kg dosage groups. The rabbit appears to be a very sensitive species to BOTOX treatment.
Impairment of fertility and reproduction
The reproductive NOEL following i.m. injection of BOTOX was 4 Units/kg in male rats and 8 Units/kg in female rats. Higher dosages were associated with dose-dependent reductions in fertility. Provided impregnation occurred, there were no adverse effects on the numbers or viability of the embryos sired or conceived by treated male or female rats.
Pre- and post-natal developmental effects
In female rats, the reproductive NOEL was 16 Units/kg. The developmental NOEL was 4 Units/kg.
Antigenicity
BOTOX showed antigenicity in mice only in the presence of adjuvant. BOTOX was found to be slightly antigenic in the guinea pig.
Blood compatibility
No haemolysis was detected up to 100 Units/ml of BOTOX in normal human blood.
6 PHARMACEUTICAL PARTICULARS 6.1 List of excipients
Human albumin Sodium chloride
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product should not be mixed with other medicinal products.
6.3 Shelf life
3 years.
After reconstitution, stability has been demonstrated for 24 hours at 2°C - 8°C. From a microbiological point of view, the product should be used immediately. If not used immediately, in-use storage times and conditions prior to use are the responsibility of the user and would normally not be longer than 24 hours at 2°C to 8°C (see also section 6.6).
6.4 Special precautions for storage
Store in a refrigerator (2°C-8°C), or store in a freezer (at or below -5°C).
For storage conditions of the reconstituted medicinal product see section 6.3.
6.5 Nature and contents of container
Clear glass vial, with rubber stopper and tamper-proof aluminium seal, containing white powder for solution for injection.
Pack size:
• Carton comprising one 200 Allergan Unit vial and package leaflet.
• Packs containing one, two, three or six cartons.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal
Reconstitution
BOTOX is reconstituted prior to use with sterile 9 mg/ml (0.9%) preservative-free sodium chloride solution for injection. It is good practice to perform vial reconstitution and syringe preparation over plastic-lined paper towels to catch any spillage. An appropriate amount of diluent (see dilution table below) is drawn up into a syringe. The exposed portion of the rubber septum of the vial is cleaned with alcohol (70%) prior to insertion of the needle. Since BOTOX is denatured by bubbling or similar violent agitation, the diluent should be injected gently into the vial. Discard the vial if a vacuum does not pull the diluent into the vial.
Reconstituted BOTOX is a clear colourless to slightly yellow solution free of particulate matter. When reconstituted, BOTOX may be stored in a refrigerator (2-8°C) for up to 24 hours prior to use. After this period used or unused vials should be discarded.
Each vial is for single use only.
Care should be taken to use the correct diluent volume for the presentation chosen to prevent accidental overdose. If different vial sizes of BOTOX are being used as part of one injection procedure, care should be taken to use the correct amount of diluent when reconstituting a particular number of units per 0.1 ml. The amount of diluent varies between BOTOX 50 Allergan Units, BOTOX 100 Allergan Units and BOTOX 200 Allergan Units. Each syringe should be labelled accordingly.
Dilution table for BOTOX 50, 100 and 200 Allergan Units vial size for all
|
indications exce |
pt bladder disorders: | ||
|
50 Unit vial |
100 Unit vial |
200 Unit vial | |
|
Resulting |
Amount of diluent |
Amount of diluent |
Amount of diluent |
|
dose (Units per 0.1 ml) |
(sodium chloride 9 mg/ml (0.9%) solution for injection) added in a 50 Unit vial |
(sodium chloride 9 mg/ml (0.9%) solution for injection) added in a 100 Unit vial |
(sodium chloride 9 mg/ml (0.9%) solution for injection) added in a 200 Unit vial |
|
20 Units |
0.25 ml |
0.5 ml |
1 ml |
|
10 Units |
0.5 ml |
1 ml |
2 ml |
|
5 Units |
1 ml |
2 ml |
4 ml |
|
4 Units |
1.25 ml |
2.5 ml |
5 ml |
|
2.5 Units |
2 ml |
4 ml |
8 ml |
|
1.25 Units |
4 ml |
8 ml |
N/A |
Overactive bladder:
It is recommended that a 100 Unit or two 50 Unit vials are used for convenience of reconstitution.
Dilution instructions using two 50 Unit vials:
• Reconstitute two 50 Unit vials of BOTOX each with 5 ml of 9 mg/ml (0.9%) preservative-free sodium chloride solution and mix the vials gently.
• Draw the 5 ml from each of the vials into a single 10 ml syringe.
This will result in a 10 ml syringe containing a total of 100 Units of reconstituted BOTOX. Use immediately after reconstitution in the syringe. Dispose of any unused saline.
Dilution instructions using a 100 Unit vial:
• Reconstitute a 100 Unit vial of BOTOX with 10 ml of 9 mg/ml (0.9%) preservative-free sodium chloride solution and mix gently.
• Draw the 10 ml from the vial into a 10 ml syringe.
This will result in a 10 ml syringe containing a total of 100 Units of reconstituted BOTOX. Use immediately after reconstitution in the syringe. Dispose of any unused saline.
Dilution instructions using a 200 Unit vial:
• Reconstitute a 200 Unit vial of BOTOX with 8 ml of 9 mg/ml (0.9%) preservative-free sodium chloride solution and mix gently.
• Draw 4 ml from the vial into a 10 ml syringe.
• Complete the reconstitution by adding 6 ml of 9 mg/ml (0.9%) preservative-free sodium chloride solution into the 10 ml syringe and mix gently.
This will result in a 10 ml syringe containing a total of 100 Units of reconstituted BOTOX. Use immediately after reconstitution in the syringe. Dispose of any unused saline.
This product is for single use only and any unused reconstituted product should be disposed of.
Urinary incontinence due to neurogenic detrusor overactivity:
It is recommended that a 200 Unit vial or two 100 Unit vials are used for convenience of reconstitution.
Dilution instructions using four 50 Unit vials:
• Reconstitute four 50 Unit vials of BOTOX, each with 3 ml of 9 mg/ml (0.9%) preservative-free sodium chloride solution for injection and mix the vials gently.
• Draw 3 ml from the first vial and 1 ml from the second vial into one 10 ml syringe.
• Draw 3 ml from the third vial and 1 ml from the fourth vial into a second 10 ml syringe.
• Draw the remaining 2 ml from the second and fourth vials into a third 10 ml syringe.
• Complete the reconstitution by adding 6 ml of 9 mg/ml (0.9%) preservative-free sodium chloride solution for injection into each of the three 10 ml syringes, and mix gently.
This will result in three 10 ml syringes containing a total of 200 Units of reconstituted
BOTOX. Use immediately after reconstitution in the syringe. Dispose of any unused
saline.
Dilution instructions using two 100 Unit vials:
• Reconstitute two 100 Unit vials of BOTOX, each with 6 ml of 9 mg/ml (0.9%) preservative-free sodium chloride solution for injection and mix the vials gently.
• Draw 4 ml from each vial into each of two 10 ml syringes.
• Draw the remaining 2 ml from each vial into a third 10 ml syringe.
• Complete the reconstitution by adding 6 ml of 9 mg/ml (0.9%) preservative-free sodium chloride solution for injection into each of the 10 ml syringes, and mix gently.
This will result in three 10 ml syringes containing a total of 200 Units of reconstituted
BOTOX. Use immediately after reconstitution in the syringe. Dispose of any unused
saline.
Dilution instructions using a 200 Unit vial:
• Reconstitute a 200 Unit vial of BOTOX with 6 ml of 9 mg/ml (0.9%) preservative-free sodium chloride solution for injection and mix the vials gently.
• Draw 2 ml from the vial into each of three 10 ml syringes.
• Complete the reconstitution by adding 8 ml of 9 mg/ml (0.9%) preservative-free sodium chloride solution for injection into each of the 10 ml syringes, and mix gently.
This will result in three 10 ml syringes containing a total of 200 Units of reconstituted BOTOX. Use immediately after reconstitution in the syringe. Dispose of any unused saline.
The 'unit' by which the potency of preparations of BOTOX is measured should be used to calculate dosages of BOTOX only and is not transferable to other preparations of botulinum toxin.
Disposal
For safe disposal, unused vials should be reconstituted with a small amount of water then autoclaved. Any used vials, syringes, and spillages etc. should be autoclaved, or the residual BOTOX inactivated using dilute hypochlorite solution (0.5%).
Any unused product or waste material should be disposed of in accordance with local requirements.
Allergan Limited, Marlow International, The Parkway, Marlow, Bucks, SL7 1YL, UK
8 MARKETING AUTHORISATION NUMBER(S)
PL 00426/0119
9 DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
30/03/2009
10 DATE OF REVISION OF THE TEXT
19/03/2015