Breakyl 400 Microgram Buccal Film
Out of date information, search anotherSUMMARY OF PRODUCT CHARACTERISTICS
1 NAME OF THE MEDICINAL PRODUCT
BREAKYL 400 microgram buccal film
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
Breakyl 400 microgram buccal film One buccal film contains:
400 micrograms fentanyl (as fentanyl citrate),
Excipients with known effect:
|
Breakyl, strength (microgram) Excipient |
400 |
|
propylene glycol (E1520) (mg) |
0.35 |
|
sodium benzoate (E211) (mg) |
0.23 |
|
methyl parahydroxybenzoate (E218) (mg) |
0.24 |
|
propyl parahydroxybenzoate (E216) (mg) |
0.06 |
For the full list of excipients, see section 6.1
3 PHARMACEUTICAL FORM
Buccal film
Breakyl is a soluble rectangular, flat, flexible buccal film with a pink side and a white side designed to deliver fentanyl directly into the blood circulation.
The pink side contains the active substance fentanyl. The white side minimises fentanyl release into the saliva to avoid swallowing of the active substance.
The following stencil shows the sizes of the available Breakyl strengths:

2DC 4DC 60C 8CG 12DC
microgam microgram microgram microgram microgram
0.78 cm2 1.56 cm2 2.34 cm2 3.11 cm2 4.67 cm2
Each buccal film is individually sealed in a child resistant sachet.
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
Breakyl is indicated for the treatment of breakthrough pain (BTP) in adults with cancer who are already receiving maintenance opioid therapy for chronic cancer pain.
BTP is a transitory exacerbation of pain that occurs on a background of otherwise controlled persistent pain.
Patients receiving maintenance opioid therapy are those who are taking at least 60 mg of oral morphine daily, at least 25 micrograms of transdermal fentanyl per hour, at least 30 mg of oxycodone daily, at least 8 mg of oral hydromorphone daily or an equi-analgesic dose of another opioid for a week or longer.
4.2 Posology and method of administration
Treatment has to be initiated by and maintained under the guidance of a physician experienced in the management of opioid therapy in cancer patients. In order to minimise the risks of opioid-related undesirable effects and to identify the “successful” dose, it is imperative that patients be monitored closely by health professionals during the titration process.
As the successful dose of Breakyl for breakthrough cancer pain cannot be predicted from the daily maintenance dose of opioids or from other medication for breakthrough cancer pain, it has to be determined by dose titration.
Dose Titration
Before patients start titration with Breakyl, it is expected that their background persistent pain is controlled by maintenance use of opioid therapy. In case patients experience more than four breakthrough pain episodes per day, increasing the maintenance opioid dose has to be considered before starting the titration process with Breakyl.
Titration in patients switching from other fentanyl containing products Due to different absorption profiles, switching must not be done at a 1:1 ratio. If switching from another oral fentanyl citrate product, independent dose titration with Breakyl is required as bioavailability between products differs significantly (see graph under section 5.2).
Starting Dose:
The initial dose of Breakyl should be 200 micrograms, titrating upwards as necessary using the range of available dosage strengths (200, 400, 600, 800, 1200 micrograms). Titration process should be carefully monitored until a dose is reached that provides adequate analgesia with acceptable undesirable effects after a single dose per episode of breakthrough pain. This is defined as the successful dose. Doses of Breakyl must be separated by at least 4 hours.
Two presentations of Breakyl are available for dose titration:
Breakyl Start, containing one buccal film of 200, 400, 600, and 800 microgram each and Breakyl 200 microgram, buccal film.
With Breakyl 200 microgram, higher doses may be achieved by applying a combination of Breakyl 200 microgram buccal films simultaneously:
1 buccal film Breakyl 200 equals a dose of 200 micrograms
2 buccal films Breakyl 200 equal a dose of 400 micrograms
3 buccal films Breakyl 200 equal a dose of 600 micrograms
4 buccal films Breakyl 200 equal a dose of 800 micrograms
In case the highest strength in Breakyl Start (800 micrograms) or the combination of 4 buccal films Breakyl 200 at one time (800 micrograms) is not sufficient for pain relief, Breakyl 1200 may be indicated. This is the highest available strength of Breakyl.
If adequate pain relief is achieved after application of a particular dose strength, subsequent breakthrough pain episodes should be treated by using the identified Breakyl dose.
If adequate pain relief is not achieved within 30 minutes after application of a particular Breakyl dose, and the patient has tolerated the dose, the patient should treat the subsequent breakthrough pain episode by using the next higher Breakyl dose.
If adequate pain relief is not achieved within 30 minutes after application of the 1200 microgram Breakyl buccal film, (the highest available dose strength), the patient should discuss treatment options with their physician. The combined use of low dose buccal films to reach the next higher dose is possible during titration. The combined use of total doses exceeding 1200 micrograms has not been evaluated under controlled conditions.
During any episode of breakthrough pain, if adequate pain relief is not achieved within 30 minutes after application of a Breakyl buccal film, the patient may use a rescue medicinal product for breakthrough pain, if directed by their physician. However, opioid rescue medicinal products should not be used when unacceptable undesirable effects of Breakyl or signs of opioid toxicity have been noted.
BREAKYL is available in 5 dosage strengths; 200, 400, 600, 800 and 1200 gg

Doses of BREAKYL must be separated by at least 4 hours. During any episode of breakthrough pain, if adequate pain relief is not achieved within 30 minutes, the patient may use a rescue medication as directed.
Maintenance Therapy
Once a successful dose is determined, usage of Breakyl should be limited to four or fewer episodes of breakthrough pain per day, which must be separated by at least 4 hours. Breakyl should only be used once per episode.
Dose Readjustment
Dosage readjustment of either Breakyl or the maintenance (around-the-clock) opioid analgesic may be required in some patients in order to continue to provide adequate relief of breakthrough pain. Increasing the around-the-clock opioid dose used for persistent pain should be considered in patients experiencing more than four breakthrough pain episodes daily over a period of more than four consecutive days. If the dose of the long acting opioid is increased, the dose of Breakyl to treat breakthrough pain may need to be reviewed.
It is imperative that any dose re-titration of any analgesic is monitored by a physician.
Discontinuation of therapy
Provided patients continue to take their chronic opioid therapy for persistent pain, Breakyl therapy may usually be immediately discontinued if no longer required for breakthrough pain only.
For patients requiring discontinuation of all opioid therapy, account should be taken of the Breakyl dose in consideration of a gradual downward opioid titration to avoid the possibility of abrupt withdrawal effects.
Application of Breakyl The patient should:
- open the Breakyl sachet immediately prior to use as indicated by the instructions printed on the sachet;
- use their tongue to wet the inside of their cheek or rinse their mouth with water to moisten the area for placement of Breakyl;
- with dry hands, take the Breakyl buccal film between forefinger and thumb with the pink side facing to the thumb;
- place the Breakyl buccal film inside his/ her mouth, so that the pink side makes smooth contact with the inner lining of his/ her cheek
- press and hold it in place for a minimum of 5 seconds until it sticks firmly; then the white side should be visible.
The Breakyl buccal film should stay in place on its own after this period. Liquids may be consumed after 5 minutes.
The Breakyl buccal film will usually dissolve completely within 15 to 30 minutes after application. In individual cases complete dissolution of the product may take more than thirty minutes, but this does not affect fentanyl absorption. The patient should be instructed to avoid manipulating the buccal film with their tongue or finger(s) and avoid eating food until the buccal film has dissolved.
A buccal film of Breakyl, if chewed and swallowed, might result in lower peak concentrations and lower bioavailability than when used as directed (see section 5.2).
Use in children and adolescents:
Breakyl is not recommended for use in children and adolescents below 18 years due to a lack of data on safety and efficacy.
Use in the elderly:
Elderly patients have been shown to be more sensitive to the effects of fentanyl when administered intravenously compared with the younger population. In the elderly, elimination of fentanyl is slower and the terminal elimination half-life is longer, which may result in accumulation of the active substance and a greater risk of undesirable effects. Therefore dose titration needs to be approached with particular care. However, in clinical studies, there was no difference in the median titrated dose of Breakyl on patients aged 65 years and older compared to those < 65 years.
Use in special patient populations:
Special care should be taken during the titration process in patients with kidney or liver dysfunction.
Patients with Grade 1 mucositis should be monitored closely, dose adjustment may be considered. The efficacy and safety of Breakyl in patients with mucositis more severe than Grade1 have not been studied.
The buccal film must not be used if the sachet has been damaged before opening.
4.3 Contraindications
Hypersensitivity to fentanyl or to any of the excipients (see section 6.1).
Simultaneous use of monoamine-oxidase (MAO) inhibitors, or within 2 weeks after the cessation of the use of MAO inhibitors (see also section 4.5).
Severe respiratory depression or severe obstructive lung conditions.
Patients without maintenance opioid therapy (see section 4.1) as there is an increased risk of respiratory depression.
4.4 Special warnings and precautions for use
Patients and their caregivers must be instructed that Breakyl contains an active substance in an amount which can be fatal to a child, and therefore to keep Breakyl out of reach and sight of children and non-patients at all times.
In order to minimise the risks of opioid-related undesirable effects and to identify the effective dose, it is imperative that patients be monitored closely by a physician during the titration process.
It is important that the long acting opioid treatment used to treat the patient's persistent pain has been stabilised before Breakyl therapy begins.
There is a risk of clinically significant respiratory depression associated with the use of Breakyl. Particular caution should be used when titrating Breakyl in patients with non-severe chronic obstructive pulmonary disease or other medical conditions predisposing them to respiratory depression, as even normally therapeutic doses of Breakyl may further decrease respiratory drive to the point of respiratory failure.
Breakyl should only be administered with extreme caution in patients who may be particularly susceptible to the intracranial effects of CO2 retention, such as those with evidence of increased intracranial pressure, or impaired consciousness. Opioids may obscure the clinical course of a patient with a head injury and should be used only if clinically warranted.
Intravenous fentanyl may produce bradycardia. Therefore, Breakyl should be used with caution in patients with bradyarrhythmias.
Careful consideration should be given to patients with hypovolaemia and hypotension.
In addition, Breakyl should be administered with caution to patients with liver or kidney dysfunction. The influence of hepatic or renal impairment on the pharmacokinetics of the medicinal product has not been evaluated. However, when administered intravenously, the clearance of fentanyl has been shown to be altered in hepatic and renal disease due to alterations in metabolic clearance and plasma proteins. After administration of Breakyl, impaired liver or renal function may both increase the bioavailability of fentanyl and decrease its systemic clearance, which could lead to increased and prolonged opioid effects. Therefore, special care should be taken during the titration process in patients with moderate or severe hepatic or renal disease.
Tolerance and physical and/or psychological dependence may develop upon repeated administration of opioids such as fentanyl. However, iatrogenic addiction following therapeutic use of opioids is rare.
Fentanyl contained in this medicinal product could produce a positive analytic result in anti-doping tests.
Breakyl contains sodium benzoate, methyl-parahydroxybenzoate, propyl-parahydroxybenzoate, and propylene glycol. Sodium benzoate is mildly irritant to the skin, eyes and mucous membranes. Methyl-parahydroxybenzoate and propyl-parahydroxybenzoate may cause allergic reactions (possibly delayed). Propylene glycol may cause skin irritation.
Patients with Grade 1 mucositis should be monitored closely, dose adjustment may be considered. The efficacy and safety of Breakyl in patients with mucositis more severe than Grade1 have not been studied (see also sections 4.2 and 5.2).
4.5 Interaction with other medicinal products and other forms of interaction
Breakyl should not be used in patients who are receiving or have received monoamine oxidase (MAO) inhibitors within the last 14 days because severe and unpredictable potentiation by MAO inhibitors has been reported with opioid analgesics (see section 4.3).
Fentanyl is metabolised by the CYP3A4 isoenzyme in the liver and intestinal mucosa (please see also section 5.2). Inhibitors of CYP3A4 such as
- macrolide antibiotics (e.g. erythromycin, clarithromycin, telithromycin),
- azole antifungals (e.g. ketoconazole, itraconazole, and fluconazole),
- certain protease inhibitors (e.g. ritonavir, indinavir, nelfinavir, saquinavir),
- calcium channel blockers (e.g. diltiazem or verapamil),
- anti-emetics (e.g. aprepitant or dronabinol),
- antidepressants (e.g. fluoxetine),
- antacids (e.g. cimetidine), or
caffeine and alcohol, may increase the bioavailability of swallowed fentanyl and may also decrease its systemic clearance which may result in increased or prolonged opioid effects and may cause potentially fatal respiratory depression. Similar effects could be seen after concurrent ingestion of grapefruit juice, which is known to inhibit CYP3A4. Hence caution is advised if fentanyl is given concomitantly with CYP3A4 inhibitors. Patients receiving Breakyl who begin therapy with, or increase the dose of, CYP3A4 inhibitors should be carefully monitored for signs of opioid toxicity over an extended period of time.
The concomitant use of Breakyl with potent CYP3A4 inducers such as
- barbiturates and other sedatives (e.g.,phenobarbital),
- anti-epileptics (e.g. carbamazepine, phenytoin, oxcarbazepine),
- certain antivirals (e.g. efavirenz, nevirapine),
- anti-inflammatory or immunosuppressive agents (e.g. glucocorticoids),
- anti-diabetics (e.g. pioglitazone),
- antibiotics for tuberculosis treatment (e.g. rifabutin, rifampin),
- psychotropic substances (e.g. modafinil),
- antidepressants (e.g. St. John's wort), or
nicotin and alcohol, may result in a decrease in fentanyl plasma concentrations, which could decrease the efficacy of Breakyl. Patients receiving Breakyl who stop therapy with, or decrease the dose of CYP3A4 inducers should be monitored for signs of increased Breakyl activity or toxicity and the dose of Breakyl should be adjusted accordingly.
The concomitant use of other CNS depressants, including other opioids, sedatives or hypnotics, general anaesthetics, phenothiazines, tranquillisers, skeletal muscle relaxants, sedating antihistamines and alcohol may produce additive depressant effects.
Withdrawal symptoms may be precipitated through the administration of active substances with opioid antagonist activity, e.g. naloxone, or partial agonist analgesics (e.g. pentazocine, butorphanol, buprenorphine, nalbuphine).
4.6 Fertility, pregnancy and lactation
There are no adequate data from the use of fentanyl in pregnant women. Studies in animals have shown reproductive toxicity (see section 5.3).The potential risk for humans is unknown. Fentanyl should not be used in pregnancy unless clearly necessary.
Following long-term treatment, fentanyl may cause withdrawal symptoms in the new-born infant.
It is advised not to use fentanyl during labour and delivery (including caesarean section) because fentanyl passes through the placenta and may cause respiratory depression in the foetus. If Breakyl is administered, an antidote for the child should be readily available.
Fentanyl passes into breast milk and may cause sedation and respiratory depression in the breast-fed child. Fentanyl should not be used by breastfeeding women and breast-feeding should not be restarted until at least 48 hours after the last administration of fentanyl.
4.7 Effects on ability to drive and use machines
No studies of the effects on the ability to drive and use machines have been performed.
However, opioid analgesics may impair the mental and/or physical ability required for the performance of potentially dangerous tasks (e.g., driving a car or operating machinery). Patients should be advised not to drive or operate machinery if they experience somnolence, dizziness, blurred or double vision while taking Breakyl.
This medicine can impair cognitive function and can affect a patient’s ability to drive safely. This class of medicine is in the list of drugs included in regulations under 5a of the Road Traffic Act 1988. When prescribing this medicine, patients should be told:
• The medicine is likely to affect your ability to drive
• Do not drive until you know how the medicine affects you
• It is an offence to drive while under the influence of this medicine
• However, you would not be committing an offence (called ‘statutory defence’) if:
- The medicine has been prescribed to treat a medical or dental problem and
- You have taken it according to the instructions given by the prescriber and in the information provided with the medicine and
- It was not affecting your ability to drive safely
4.8 Undesirable effects
Typical opioid undesirable effects are to be expected with Breakyl. Frequently, these will cease or decrease in intensity with continued use of the product, as the patient is titrated to the most appropriate dose. The most serious adverse reactions associated with all opioids including Breakyl are respiratory depression (potentially leading to respiratory arrest), circulatory depression, hypotension and shock, and all patients should be closely monitored for these.
Because the clinical studies of Breakyl were designed to evaluate safety and efficacy in treating patients with breakthrough pain associated with cancer, all patients were also taking concomitant opioids, such as sustained-release morphine, sustained-release oxycodone or transdermal fentanyl, for their persistent pain. Thus, it is not possible to definitively separate the effects of Breakyl alone.
The adverse reaction data presented here reflect on the one hand the current experience with Breakyl for breakthrough pain along with a concomitant opioid for persistent pain. On the other hand, adverse events listed as very rare have previously been associated with the substance fentanyl, but have not been observed during clinical studies with Breakyl up to now. There has been no attempt to correct for concomitant use of other opioids, duration of Breakyl therapy, or cancer-related symptoms.
The most frequent adverse reactions observed were nausea, somnolence, and dizziness.
Evaluation of undesirable effects is based on the following frequencies: very common (>1/10), common (>1/100 to <1/10), uncommon (>1/1,000 to <1/100), rare (>1/10,000 to <1/1,000), very rare (<1/10,000) or not known (cannot be estimated from the available data).
The adverse events considered to be at least possibly-related to treatment were as follows:
Metabolism and nutrition disorders
Uncommon: anorexia
Psychiatric disorders
Common: confusional state
Uncommon: anxiety, hallucination, delusion, abnormal dreams, nervousness, insomnia, restlessness
Very rare: abnormal thinking, depersonalisation, depression, emotional lability, euphoria
Nervous system disorders
Common: somnolence, dizziness, headache, sedation
Uncommon: dysgeusia, lethargy, amnesia, cognitive disorder
Very rare: myoclonus, paraesthesia (including hyperaesthesia/ circumoral
paraesthesia), abnormal gait/ incoordination
Eye disorders
Common: abnormal vision (blurred, diplopia)
Vascular disorders
Uncommon: hot flush Very rare: vasodilatation
Respiratory, thoracic and mediastinal disorders
Uncommon: respiratory depression, sinus congestion Very rare: dyspnoea
Gastrointestinal disorders
Common: nausea, constipation, vomiting, dry mouth
Uncommon: diarrhoea, stomatitis, gingival bleeding, dyspepsia, mouth
ulceration, oral pain, odynophagia
Very rare: abdominal pain, flatulence, abdomen enlarged
Skin and subcutaneous tissue disorders
Common: pruritus
Uncommon: hyperhidrosis, increased tendency to bruise Very rare: rash
Musculoskeletal and connective tissues disorders
Uncommon: muscle twitching, arthralgia, muscular weakness, musculoskeletal pain, pain in extremity, pain in jaw
Renal and urinary disorders
Uncommon: urinary incontinence Very rare: urinary retention
General disorders and administration site conditions
Common: fatigue
Uncommon: asthenia, chills, pyrexia, thirst Very rare: malaise
Investigations
Uncommon: blood pressure increased
Injury, poisoning and procedural complications
Uncommon: accidental injury (for example, falls)
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Yellow Card Scheme at: www.mhra.gov.uk/yellowcard.
4.9 Overdose
The symptoms of Breakyl overdose are expected to be similar in nature to those of intravenous fentanyl and other opioids, and are an extension of its pharmacological actions, with the most serious significant effect being respiratory depression.
Immediate management of opioid overdose includes removal of the Breakyl buccal film, if still in the mouth, ensuring a patent airway, physical and verbal stimulation of the patient, assessment of the level of consciousness, ventilatory and circulatory status, and assisted ventilation (ventilatory support) if necessary.
For treatment of accidental ingestion in the opioid naive person, intravenous access should be obtained, and naloxone or other opioid antagonists should be employed as clinically indicated. The duration of respiratory depression following overdose may be longer than the effects of the opioid antagonist's action (e.g., the half-life of naloxone ranges from 30 to 81 minutes) and repeated administration of naloxone or other opioid antagonists may be necessary. The Summary of Product Characteristics of the individual opioid antagonist should be consulted for details about such use.
For treatment of overdose in opioid-maintained patients, intravenous access should be obtained. The judicious use of naloxone or another opioid antagonist may be warranted in some instances, but it is associated with the risk of precipitating an acute withdrawal syndrome.
Although muscle rigidity interfering with respiration has not been seen following the use of Breakyl, this is possible with fentanyl and other opioids.
If it occurs, it should be managed by the use of assisted ventilation, by an opioid antagonist, and as a final alternative, by a neuromuscular blocking agent.
5 PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: analgesics; opioids, phenylpiperidine derivatives ATC code: N02AB03
Fentanyl, a pure opioid agonist, acts primarily through interaction with p-opioid receptors located in the brain, spinal cord and smooth muscle. The primary site of therapeutic action is the central nervous system (CNS). The clinically most useful pharmacological effect of the interaction of fentanyl with p-opioid receptors is analgesia.
In patients with chronic cancer pain on stable doses of regularly scheduled opioids to control their persistent pain, Breakyl significantly reduced pain intensity, assessed as the sum of pain intensity differences (SPID), compared to placebo during 15, 30, 45, 60 min following administration.
The analgesic effects of fentanyl are related to the blood level of the active substance, if proper allowance is made for the delay into and out of the CNS (a process with a 3-5 minute half-life). In opioid-naive individuals, analgesia occurs at blood levels of 1 to 2 ng/ml, while blood levels of 10-20 ng/ml would produce surgical anaesthesia and profound respiratory depression.
Secondary actions include increase in the tone and decrease in the contractions of the gastrointestinal smooth muscle, which results in prolongation of gastrointestinal transit time and may be responsible for the constipation effect of opioids.
While opioids generally increase the tone of urinary tract smooth muscle, the overall effect tends to vary, in some cases producing urinary urgency, in others difficulty in urination.
All opioid p-receptor agonists, including fentanyl, produce dose dependent respiratory depression. The risk of respiratory depression is less in patients with pain receiving chronic opioid therapy who develop tolerance to respiratory depression and other opioid effects.
5.2 Pharmacokinetic properties
General introduction
Fentanyl is highly lipophilic and can be absorbed very rapidly through the oral mucosa and more slowly by the conventional gastrointestinal route. It is subject to first-pass hepatic and intestinal metabolism and the metabolites do not contribute to fentanyl's therapeutic effects.
Absorption
In a pharmacokinetic study, following buccal application, Breakyl was rapidly absorbed and the absolute bioavailability was 71 %. This absolute bioavailability study also demonstrated similar pharmacokinetics in the subsets of 6 male and 6 female adult normal volunteers.
The absorption pharmacokinetics of fentanyl from Breakyl is a combination of an initial rapid absorption from the buccal mucosa and a more prolonged absorption of swallowed fentanyl from the GI tract. Based on the absolute bioavailability study, approximately 51 % of the total dose of Breakyl is rapidly absorbed from the buccal mucosa and becomes systemically available. The remaining 49 % of the total dose is swallowed with the saliva and then slowly absorbed from the GI tract. About 1/3 of this amount (20 % of the total dose) escapes hepatic and intestinal first-pass elimination and becomes systemically available. Thus, the observed 71 % absolute bioavailability of Breakyl is divided between rapid transmucosal and slower GI absorption. A unit dose of Breakyl, if chewed and swallowed, will likely result in lower peak concentrations and lower bioavailability than when consumed as directed.
Dose proportionality across the available range of dosages (200 to 1200 micrograms) of Breakyl has been demonstrated. After consumption of a single buccal film of Breakyl (200 to 1200 micrograms), mean Cmax usually ranges from 0.38 to 2.19 ng/ml (depending on dose) and Tmax ranges from 45 - 240 minutes (median 60 min). Application of Breakyl on an active site of mucositis (Grade 1) in a group of cancer patients was associated with decreases in the Cmax and AUCinf. It is recommended that patients with Grade 1 mucositis are monitored closely, dose adjustment may be considered. The efficacy and safety of Breakyl when used in patients with mucositis more severe than Grade1 have not been studied.
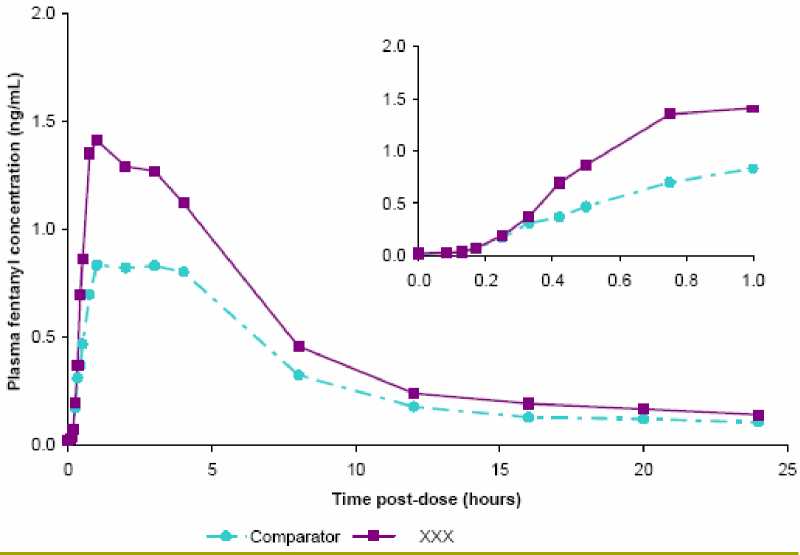
Mean fentanyl plasma concentration versus time profiles (24 hours and 1st hour only) following single doses of 800 pg BREAKYL (XXX) or 800 pg comparator (fentanyl oromucosal applicator) in healthy adult subjects
Distribution
Fentanyl is highly lipophilic. Animal data showed that following absorption, fentanyl is rapidly distributed to the brain, heart, lungs, kidneys and spleen followed by a slower redistribution to muscles and fat. The plasma protein binding of fentanyl is 80-85%. The main binding protein is alpha-1-acid glycoprotein, but both albumin and lipoproteins contribute to some extent. The free fraction of fentanyl increases with acidosis. The mean volume of distribution at steady state (Vss) is 4 l/kg.
Biotransformation
Fentanyl is metabolised in the liver and in the intestinal mucosa to norfentanyl by cytochrome P450, CYP3A4, isoform. Norfentanyl was not found to be pharmacologically active in animal studies. Fentanyl is primarily (more than 90 %) eliminated by biotransformation to N-dealkylated and hydroxylated inactive metabolites.
Elimination
Less than 7 % of the dose is excreted unchanged in the urine, and only about 1 % is excreted unchanged in the faeces. The metabolites are mainly excreted in the urine, while faecal excretion is less important. The total plasma clearance of fentanyl is 0.5 l/hr/kg (range 0.3 to 0.7 l/hr/kg). The clinically relevant half life of fentanyl after Breakyl administration is approximately seven hours, and the terminal elimination half-life is about 14 hours.
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity and carcinogenicity.
Embryo-foetal developmental toxicity studies conducted in rats and rabbits revealed no compound induced malformations or developmental variations when administered during the period of organogenesis. In a fertility and early embryonic development study in rats, a male-mediated effect was observed at high doses (300 mcg/kg/day, s.c.) and is consistent with the sedative effects of fentanyl in animal studies. In studies on pre and postnatal development in rats the survival rate of offspring was significantly reduced at doses causing severe maternal toxicity. Further findings at maternally toxic doses in F1 pups were delayed physical development, sensory functions, reflexes and behaviour. These effects could either be indirect effects due to altered maternal care and/or decreased lactation rate or a direct effect of fentanyl on the pups.
Carcinogenicity studies (26-week dermal alternative bioassay in Tg.AC transgenic mice; two-year subcutaneous carcinogenicity study in rats) did not induce any findings indicative of oncogenic potential.
6 PHARMACEUTICAL PARTICULARS
6.1 List of excipients
Active layer:
propylene glycol (E1520), sodium benzoate (E211), methyl-parahydroxybenzoate (E218), propyl-parahydroxybenzoate (E216), ferric oxide (red) (E 172), anhydrous citric acid, all-rac-alpha-tocopheryl acetate, monobasic sodium phosphate (anhydrous), sodium hydroxide,
tribasic sodium phosphate (anhydrous), polycarbophil, hydroxypropyl cellulose, hydroxyethyl cellulose, carmellose sodium
Backing layer: sodium benzoate (E211), methyl-parahydroxybenzoate (E218), propyl-parahydroxybenzoate (E216),
anhydrous citric acid, all-rac-alpha-tocopheryl acetate, hydroxypropyl cellulose, hydroxyethyl cellulose, titanium dioxide (E 171), saccharin sodium, peppermint oil
6.2 Incompatibilities
Not applicable
6.3 Shelf life
2 years
6.4 Special precautions for storage
Do not store above 30 °C.
Do not refrigerate.
Store in the original package in order to protect from moisture
6.5 Nature and contents of container
Each buccal film is individually wrapped in a child-resistant sachet, consisting of a polyacrylonitrile/ aluminium/ polyamide/ paper laminate for the Aveva product.
Each buccal film is individually wrapped in a child-resistant sachet, consisting of a polyacrylonitrile/ aluminium/ polyethylene terephthalate (PET)/ paper laminate for the LTS product.
Breakyl 400 microgram buccal film:
Cartons of 3, 4, 10, 28 or 30 sachets with one buccal film each. Not all pack sizes may be marketed.
Any unused product or waste material should be disposed of in accordance with local requirements.
7
Meda Pharmaceuticals Ltd Skyway House Parsonage Road Takeley
Bishop’s Stortford CM22 6PU United Kingdom
8
9
MARKETING AUTHORISATION NUMBER(S)
PL 15142/0069
DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
31/12/2012
DATE OF REVISION OF THE TEXT
25/07/2014