Mireffik 20 Micrograms/24 Hours Intrauterine Delivery System
SUMMARY OF PRODUCT CHARACTERISTICS
1 NAME OF THE MEDICINAL PRODUCT
Mireffik 20 micrograms/24 hours Intrauterine Delivery System
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
The active substance is Levonorgestrel.
The intrauterine delivery system contains 52 mg levonorgestrel. The initial release of levonorgestrel is approximately 20 micrograms per day reducing to approximately 12 micrograms per day after 3 years.
For the full list of excipients, see section 6.1
3 PHARMACEUTICAL FORM
Levonorgestrel Intrauterine delivery system (IUS).
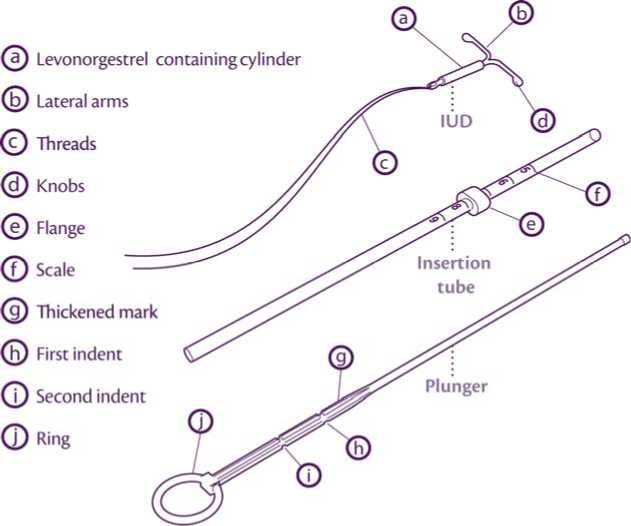
The product consists of an inserter and levonorgestrel IUS, which is loaded at the tip of the inserter. Inserter components are an insertion tube, plunger, flange, body and slider. The device consists of a white or almost white hormone-elastomer core, mounted on a T-body and covered in opaque tubing, which regulates the release of levonorgestrel. The T-body has a loop at one end and two arms at the other end. Removal threads are attached to the loop.
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
Contraception.
Heavy menstrual bleeding. Mireffik may be particularly useful in women with heavy menstrual bleeding requiring (reversible) contraception.
4.2 Posology and method of administration
Starting treatment
In women of fertile age, Mireffik is inserted into the uterine cavity within seven days of the onset of menstruation. It can be replaced by a new system at any time of the cycle.
Post-partum insertion: To reduce the risk of perforation, postpartum insertions should be postponed until the uterus is fully involuted. Do not insert earlier than six weeks after delivery. If the patient is experiencing significant post-partum bleeding and/or pain then infection or other causes should be excluded before insertion. Mireffik can also be inserted immediately after the first trimester abortion.
Mireffik is effective for three years in the indications for contraception and heavy menstrual bleeding. Therefore it should be removed after 3 years of use.
If the user wishes to continue using the same method, a new system can be inserted at the same time, in which case no additional protection is required.
Before insertion, the patient must be informed about the efficacy, risks and side-effects of Mireffik. A gynaecological examination, including examination of the breasts and exclusion of a pregnancy, should be performed. Cervical infection and sexually transmitted diseases should be excluded. The position of the uterus and the size of the uterine cavity should be determined. The instructions for insertion should be followed carefully. The patient should be re-examined six weeks after insertion and once a year thereafter, or more frequently if clinically indicated.
Paediatric population
Mireffik has not been studied in patients below 16 years of age. There is no relevant indication for the use of Mireffik before menarche.
Instructions for use and handling
Mireffik is supplied in a sterile pack which should not be opened until required for insertion. The exposed product should be handled with aseptic precautions. If the seal of the sterile package is broken, the product should be discarded (see Section 6.6 for disposal instructions).
How to insert Mireffik
It is recommended that Mireffik should only be inserted by physicians/health care professionals who are experienced in levonorgestrel IUS insertions and/or have undergone sufficient training for levonorgestrel IUS insertion.
In case of difficult insertion and/or exceptional pain or bleeding during or after insertion, please refer to section 4.4.
• Mireffik is supplied sterile having been sterilised with ethylene oxide. Do not resterilise. For single use only. Do not use if the inner package is damaged or open. Insert before the month shown on the label.
• Mireffik is inserted with the provided inserter (figure 1) into the uterine cavity by carefully following the insertion instructions.
The following insertion instruction will be provided in the box containing the IUS.
1. In women of fertile age, Mireffik is inserted within seven days of the onset of menstruation. It can be replaced by a new system at any time of the cycle.
2. It is recommended that Mireffik should only be inserted by physicians/health care professionals who have undergone sufficient training and have read carefully these instructions before Mireffik insertion.
3. Mireffik is supplied in a sterile pack. Do not use if the inner package is damaged or open.
4. Determine the position (anteversion, retroversion) and size of the uterus by a gynaecological examination. Exclude pregnancy and contraindications.
5. Place a speculum, use appropriate antiseptic solution to clean the vagina and cervix.
6. Use cervical dilators if cervical stenosis is diagnosed. Do not force to overcome resistance.
7. Grasp the cervix with a Tenaculum forceps and apply a gentle traction in order to straighten alignment of the cervical canal and uterine cavity.
8. Determine the uterine depth by hysterometry. If uterine depth is < 5.5 cm discontinue the procedure.
Introduce the plunger and the IUD in the insertion tube
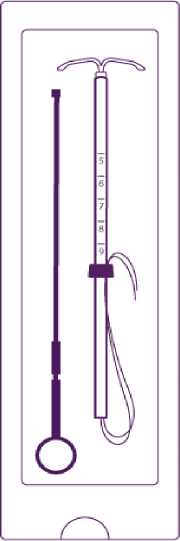
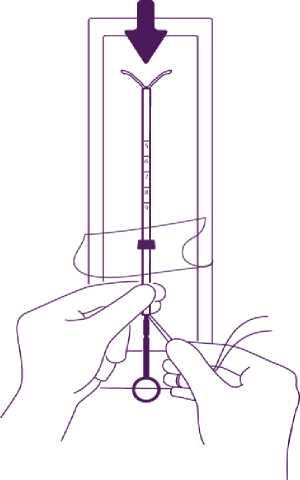
Partly open the blister (about 1/3 from the bottom) and introduce the plunger in the insertion tube. Extricate the threads from the flange. Pull the thread to introduce the IUD into the tube. The arms of the IUD must stay in an horizontal plan, parallel to the flat side of the flange.
Position the lower edge of the flange at the sounded value
Position the blue flange so as the lower edge of the flange indicates the value found by hysterometry. The flat sides of the flange must always remain parallel to the arms. This will allow the arms to open correctly in the uterine cavity.
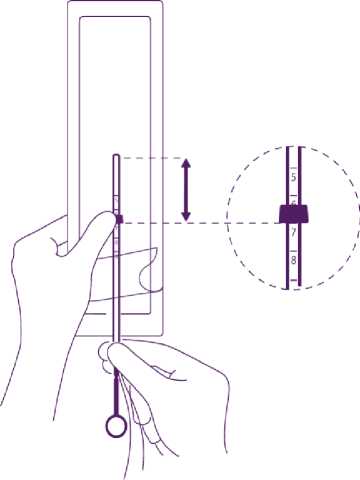
Adjust the position of the IUD in the insertion tube
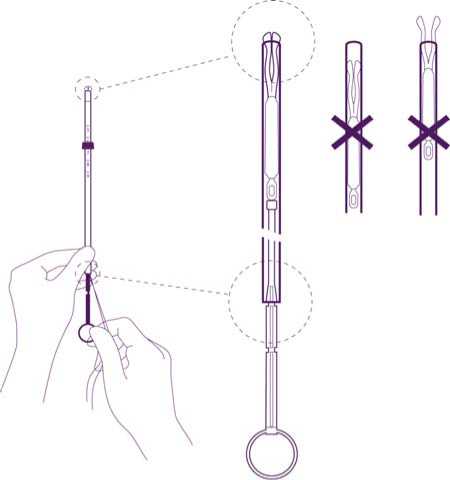
Hold the plunger firmly while pulling the thread and moving the tube to adjust the IUDs position.
The knobs of the lateral arms must be closely opposed to each other, slightly above the upper extremity of the insertion tube (see zoom 1) and the distal edge of the tube must be aligned with the first indent of the plunger (see zoom 2). If the tube is not aligned with the first indent of the plunger you must pull the thread more firmly.
Insertion
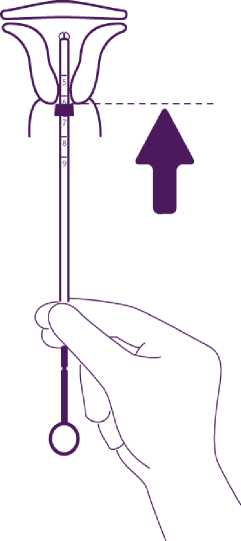
Introduce the device in the cervical canal until the blue flange is in contact with the cervix
Take the whole device out of the blister, by holding firmly the plunger and tube together in the correctly adjusted position.
Introduce the assembly into the cervical canal until the blue flange is in contact with the cervix.
Release the arms of the intrauterine device
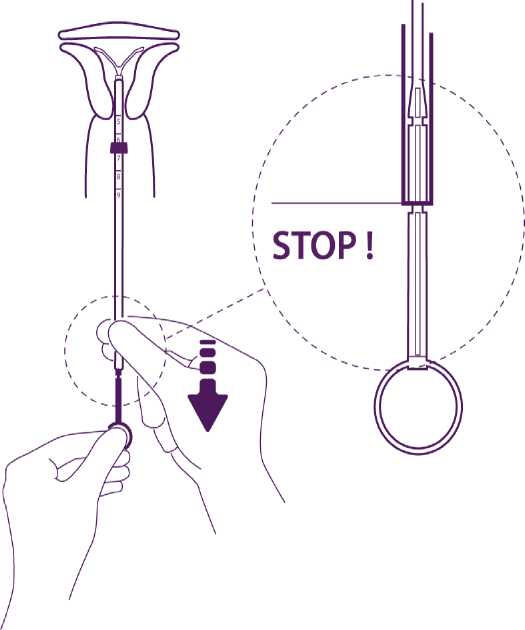
Hold the plunger, release the thread and pull the insertion tube down until its lower extremity reaches the second indent of the plunger.
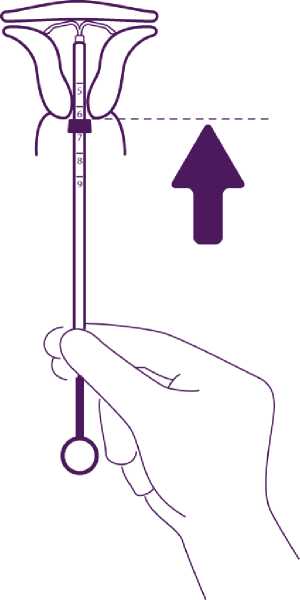
Push the IUD against the fundus
To position the IUD in the uterine cavity, push the insertion tube simultaneously with the plunger, until the blue flange is again in contact with the cervix.
Mireffik is then correctly placed in the uterine cavity.
Release the IUD from the tube into the uterine cavity
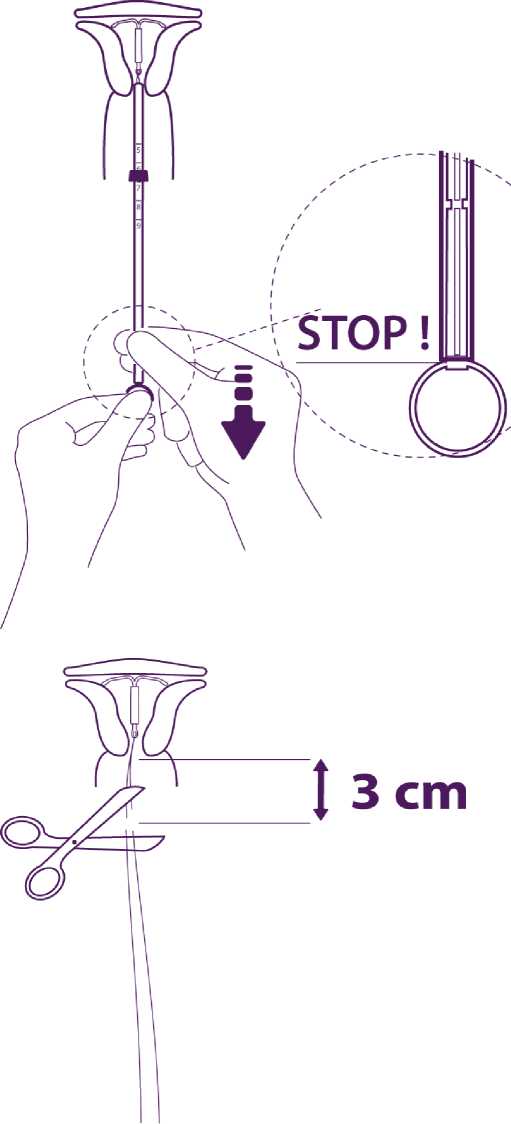
Without moving the plunger, pull the insertion tube down to the ring of the plunger.
A slight resistance marks the passage of the bulge of the plunger. Nevertheless pull down the tube to the ring of the plunger.
Mireffik is then released completely from the insertion tube.
Remove sequentially the inserter components and cut the threads
Remove sequentially, first the plunger, then the insertion tube.
Cut the threads at around 3 cm from the cervix.
IMPORTANT!
In case of difficult insertion and/or exceptional pain or bleeding during or after insertion, physical examination and ultrasound should be performed immediately to exclude perforation of the uterine body or cervix. If necessary remove the system and insert a new, sterile system.
Please report to our pharmacovigilance department any case of uterine perforation or insertion difficulties: drugsafety@mithra.com or +32 (0)4 223 38 55.
Mireffik is removed by gently pulling on the threads with forceps. If the threads are not visible and the device is in the uterine cavity, it may be removed using a narrow tenaculum. This may require dilatation of the cervical canal.
After removal of Mireffik, the device should be checked to ensure it is intact. During difficult removals, single cases have been reported of the hormone cylinder sliding over the horizontal arms and hiding them inside the cylinder. This situation does not require further intervention once completeness of the IUS has been ascertained. The knobs of the horizontal arms usually prevent complete detachment of the cylinder from the T-body.
4.3 Contraindications
• Known or suspected pregnancy
• Current or recurrent pelvic inflammatory disease
• Current genital infection
• Postpartum endometritis
• Infected abortion during the past three months
• Cervicitis, Cervical dysplasia
• Suspected or confirmed uterine or cervical malignancy
• Liver tumour or other acute or severe liver disease
• Congenital or acquired abnormality of the uterus including fibroids if they distort the uterine cavity
• Undiagnosed abnormal genital bleeding
• Conditions associated with increased susceptibility to infections
• Active or previous severe arterial disease, such as stroke or myocardial infarction
• Current or suspected hormone dependent tumours such as breast cancer (see section 4.4)
• Hypersensitivity to the active substance or to any of the excipients listed in section
6.1.
• Acute malignancies affecting the blood or leukaemias except when in remission
• Recent trophoblastic disease while hCG levels remain elevated
4.4 Special warnings and precautions for use
Medical examination
Before insertion, a complete personal and family medical history should be taken. Physical examination should be guided by this and by the contraindications and warnings for use. Pulse and blood pressure should be measured and a bimanual pelvic examination performed to establish the orientation of the uterus. The patient should be re-examined six weeks after insertion and further examinations should be performed where clinically indicated and adapted to the individual woman rather than as routine procedure. Prior to insertion pregnancy should be excluded and genital infection should be successfully treated. Women should be advised that Mireffik does not protect against HIV (AIDs) and other sexually transmitted disease (please refer to the section below on pelvic infections).
Women should be encouraged to attend cervical and breast screening as appropriate for their age.
Conditions under which Mireffik can be used with caution
Mireffik may be used with caution after specialist consultation, or removal of the system should be considered, if any of the following conditions exist or arise for the first time during treatment:
- Migraine with aura
- Unusually severe or unusually frequent headache
- Jaundice
- Marked increase of blood pressure
- Malignancies affecting the blood or leukaemias in remission
- Use of chronic corticosteroid therapy
- Past history of symptomatic functional ovarian cysts
- Active or previous severe arterial disease, such as stroke or myocardial infarction
- Severe or multiple risk factors for arterial disease
- Thrombotic arterial or any current embolic disease
- Venous thromboembolism.
In general, women using Mireffik should be encouraged to stop smoking.
Insertion / removal warnings and precautions
General Information: Insertion and removal may be associated with some pain and bleeding. In case of difficult insertion and/or exceptional pain or bleeding during or after insertion, physical examination and ultrasound should be performed immediately to exclude perforation of the uterine corpus or cervix (see also ‘Perforation’). Physical examination alone (including checking of threads) may not be sufficient to exclude partial perforation.
The procedure may precipitate fainting as a vasovagal reaction or a seizure in an epileptic patient. In the event of early signs of a vasovagal attack, insertion may need to be abandoned or the system removed. The woman should be kept supine, the head lowered and the legs elevated to the vertical position if necessary in order to restore cerebral blood flow. A clear airway must be maintained; an airway should always be at hand. Persistent bradycardia may be controlled with intravenous atropine. If oxygen is available it may be administered.
Perforation: Perforation of the uterine corpus or cervix may occur, most commonly during insertion, although it may not be detected until sometime later. This may be associated with severe pain and continued bleeding. If perforation is suspected the system should be removed as soon as possible; surgery may be required.
In a large prospective comparative non-interventional cohort study in IUS/IUD users (N = 61,448 women), the incidence of perforation was 1.3 (95% CI: 1.1 - 1.6) per 1000 insertions in the entire study cohort; 1.4 (95% CI: 1.1 - 1.8) per 1000 insertions in the Mirena cohort and 1.1 (95% CI: 0.7 - 1.6) per 1000 insertions in the copper IUD cohort.
The study showed that both breastfeeding at the time of insertion and insertion up to 36 weeks after giving birth were associated with an increased risk of perforation (see Table 1). These risk factors were independent of the type of IUS/IUD inserted.
Table 1: Incidence of perforation per 1000 insertions for the entire study cohort, stratified by breastfeeding and time since delivery at insertion (parous women)
|
Breastfeeding at time of insertion |
Not breastfeeding at time of insertion | |
|
Insertion < 36 weeks after delivery |
5.6 (95% CI 3.9-7.9; n=6047 insertions) |
1.7 (95% CI 0.8-3.1; n=5927 insertions) |
|
Insertion > 36 weeks after delivery |
1.6 (95% CI 0.0-9.1; n=608 insertions) |
0.7 (95% CI 0.5-1.1; n=41,910 insertions) |
The risk of perforation may be increased in women with a fixed retroverted uterus.
Re-examination after insertion should follow the guidance given above under the heading "Medical examination" above, which may be adapted as clinically indicated in women with risk factors for perforation.
Pelvic infection: Known risk factors for pelvic inflammatory disease are multiple sexual partners, frequent intercourse and young age. Pelvic infection may have serious consequences as it may impair fertility and increase the risk of ectopic pregnancy.
For women using Mireffik with symptoms and signs suggestive of pelvic infection appropriate antibiotics should be started. There is no need to remove Mireffik unless the symptoms fail to resolve within the following 72 hours or unless the women wishes Mireffik to be removed. Mireffik must be removed if the women experiences recurrent endometritis or pelvic infection, or if an acute infection is severe.
As with other gynaecological or surgical procedures, severe infection or sepsis (including group A streptococcal sepsis) can occur following IUS insertion, although this is extremely rare.
Complications leading to failure
Expulsion: Symptoms of the partial or complete expulsion of any IUS may include bleeding or pain. However, a system can be expelled from the uterine cavity without the woman noticing it. Partial expulsion may decrease the effectiveness of Mireffik.
As the device decreases menstrual flow, increase of menstrual flow may be indicative of an expulsion. A displaced Mireffik should be removed and a new system inserted. The woman should be advised how to check the threads of Mireffik.
Lost threads: If the retrieval threads are not visible at the cervix on follow-up examination, first exclude pregnancy. The threads may have been drawn up into the uterus or cervical canal and may reappear during the next menstrual period. If pregnancy has been excluded, the threads may usually be located by gently probing with a suitable instrument. If they cannot be found, they may have broken off, or the device may have been expelled, or rarely the device may be extrauterine after having perforated the uterus. An ultrasound should be arranged to locate the device and alternative contraception should be advised in the meantime. If an ultrasound cannot locate the device and there is no evidence of expulsion, a plain abdominal X-ray should be performed to exclude an extrauterine device.
Bleeding irregularities
Irregular bleeding: Mireffik usually achieves a significant reduction in menstrual blood loss within 3 to 6 months of treatment. Increased menstrual flow or unexpected bleeding may be indicative of expulsion. If menorrhagia persists then the woman should be re-examined. An assessment of the uterine cavity should be performed using ultrasound scan. An endometrial biopsy should also be considered.
Because irregular bleeding/spotting may occur during the first months of therapy in pre-menopausal women, it is recommended to exclude endometrial pathology before insertion of Mireffik.
When to check for pregnancy in women of child bearing potential: The possibility of pregnancy should be considered if menstruation does not occur within six weeks of the onset of previous menstruation and expulsion should be excluded. A repeated pregnancy test is not necessary in amenorrhoeic subjects unless indicated by other symptoms.
Treatment review advice for Menorrhagia: Mireffik usually achieves a significant reduction in menstrual blood loss within 3 to 6 months of treatment. If significant reduction in blood loss is not achieved in these time-frames, alternative treatments should be considered.
Other risks during use
Ectopic pregnancy : The absolute risk of ectopic pregnancy in users of levonorgestrel IUS is low. However, when a woman becomes pregnant with Mireffik in situ, the relative likelihood of ectopic pregnancy is increased. The possibility of ectopic pregnancy should be considered in the case of lower abdominal pain - especially in connection with missed periods or if an amenorrhoeic woman starts bleeding. The rate of ectopic pregnancy in users of levonorgestrel IUS is 0.06 per 100 woman-years.
This rate is lower than the rate of 0.3-0.5 per 100 woman-years estimated for women not using any contraception. The corresponding figure for the copper IUS is 0.12 per 100 woman years. Women with a previous history of ectopic pregnancy carry a higher risk of a further ectopic pregnancy.
Ovarian Cysts: Ovulatory cycles with follicular rupture usually occur in women of fertile age. Sometimes atresia of the follicle is delayed and folliculogenesis may continue. These enlarged follicles cannot be distinguished clinically from ovarian cysts. Data from clinical trials suggest that ovarian cysts have been reported as an adverse drug reaction in approximately 7% of women using Mireffik, however some published studies have reported a higher incidence of ovarian cysts (which could have been influenced by factors including frequency and criteria of ultrasound scanning, and patient population). Most of these follicles are asymptomatic, although some may be accompanied by pelvic pain or dyspareunia.
In most cases, the ovarian cysts disappear spontaneously during two to three months observation. Should this not happen, continued ultrasound monitoring and other diagnostic/therapeutic measures are recommended. Rarely, surgical intervention may be required.
Breast cancer:
A meta-analysis from 54 epidemiological studies reported that there is a slightly increased relative risk (RR = 1.24) of having breast cancer diagnosed in women who are currently using combined oral contraceptives (COCs), mainly using oestrogen-progestogen preparations. The excess risk gradually disappears during the course of the 10 years after cessation of COC use. Because breast cancer is rare in women under 40 years of age, the excess number of breast cancer diagnoses in current and recent COC users is small in relation to the overall risk of breast cancer.
The risk of having breast cancer diagnosed in users of progestogen-only methods (POPs, implants and injectables), including Mireffik, is possibly of similar magnitude to that associated with COC. However, for progestogen-only contraceptive preparations, the evidence is based on much smaller populations of users and so is less conclusive than that for COCs.
General Information
Glucose tolerance: Low-dose levonorgestrel may affect glucose tolerance and blood glucose concentrations should be monitored in diabetic users of Mireffik.
4.5 Interaction with other medicinal products and other forms of interaction
The effects of Mireffik may be impaired by drugs which induce liver enzymes, including barbiturates, primidone, phenytoin, carbamazepine, griseofulvin and rifampicin. No interaction studies on the influence of these drugs on the efficacy of Mireffik have been performed.
4.6 Fertility, Pregnancy and lactation
Pregnancy: Mireffik is not to be used during an existing or suspected pregnancy. In case of an accidental pregnancy with Mireffik in situ (see section 5: pharmacological properties), ectopic pregnancy should be excluded (see section 4.4) and the system must be removed and termination of the pregnancy should be considered as there is a high risk for pregnancy complications (abortion, infection and sepsis). Removal of Mireffik or probing of the uterus may result in spontaneous abortion. Should these procedures not be possible, the woman should be informed about these risks, and accordingly, such pregnancies should be closely monitored. The woman should be instructed to report all symptoms that suggest complications of the pregnancy, like cramping abdominal pain with fever.
Local exposure to levonorgestrel:
Clinical experience of the outcomes of pregnancies with levonorgestrel IUS in situ is limited. However, to date, there is no evidence of birth defects caused by local levonorgestrel IUS use in cases where pregnancy continues to term with the IUS in place.
Breastfeeding: Levonorgestrel is excreted in very small quantities in breast milk after use in levonorgestrel IUS. Since no risk for the child is expected, breast feeding can be continued during use of Mireffik.
Uterine bleeding has rarely been reported in women using a levonorgestrel IUS during lactation.
Fertility: the use of levonorgestrel IUS does not alter the course of female fertility after removal of the IUS.
4.7 Effects on ability to drive and use machines
Mireffik has no known influence on the ability to drive or use machines.
4.8 Undesirable effects
Undesirable effects are more common during the first months after the insertion, and subside during prolonged use.
Very common undesirable effects (occurring in more than 10% of users) include uterine/vaginal bleeding including spotting, oligomenorrhoea, amenorrhoea (see section 5.1) and benign ovarian cysts.
The frequency of benign ovarian cysts depends on the diagnostic method used, and in clinical trials enlarged follicles have been diagnosed in 12% of the subjects using a levonorgestrel IUS. Most of the follicles are asymptomatic and disappear within three months.
|
Organ system |
Undesirable effects | |||
|
Very common: >1/10 |
Common: >1/100 to <1/10 |
Uncommon: >1/1000 to <1/100 |
Rare: >1/10000 to <1/100( | |
|
Psychiatric disorders |
Depressive mood Nervousness Decreased libido |
Altered mood | ||
|
Nervous system disorders |
Headache |
Migraine | ||
|
Gastrointestinal disorders |
Abdominal pain Nausea |
Abdominal distension | ||
|
Skin and subcutaneous tissue disorders |
Acne |
Alopecia Hirsutism Pruritus Eczema |
Rash Urticaria | |
|
Musculoskeletal and connective tissue disorders |
Back pain | |||
|
Reproductive system and breast disorders |
Bleeding changes Benign ovarian cysts |
Pelvic pain Dysmenorrhoea Vaginal discharge vulvovaginitis Breast tendenress Breast pain |
Pelvic Inflammatory disease Endometritis Cervicitis Papanicolaou smear normal, class II |
Uterine perforation* |
|
General disorders and administration site conditions |
Intrauterine contraceptive device expelled |
Oedema | ||
|
Investigations |
Weight increase | |||
* This frequency is based on clinical trials that excluded breastfeeding women. In a large prospective comparative non-interventional cohort study in IUS/IUD users, the frequency of perforation in women who were breastfeeding or had an insertion up to 36 weeks after delivery was “uncommon” (see section 4.4).
Cases of sepsis (including group A streptococcal sepsis) have been reported following IUD insertion (see section 4.4).
When a woman becomes pregnant with <Product Name> in situ, the relative risk of ectopic pregnancy is increased (see ‘Special warnings and precautions for use’ and ‘Fertility, pregnancy and lactation’).
In addition, cases of breast cancer have been reported in levonorgestrel IUS users (frequency unknown, see section 4.4).
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via Yellow Card Scheme - Website: www.mhra.gov.uk/yellowcard.
4.9 Overdose
Not applicable.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Intrauterine contraceptives, plastic IUD with progestogen ATC code: G02BA03
Levonorgestrel is a progestogen used in gynaecology in various ways: as the progestogen component in oral contraceptives, in hormonal replacement therapy or alone for contraception in minipills and subdermal implants. Levonorgestrel can also be administered directly into the uterine cavity as an IUS. This allows a very low daily dosage, as the hormone is released directly into the target organ.
The contraceptive mechanism of action of the levonorgestrel IUS is based mainly on hormonal effects producing the following changes:
- Prevention of proliferation of the endometrium
- Thickening of the cervical mucus thus inhibiting the passage of sperm
- Suppression of ovulation in some women.
The physical presence of the system in the uterus would also be expected to make a minor contribution to its contraceptive effect.
When inserted according to the insertion instructions, Mireffik has a contraceptive failure rate of approximately 0.19% (95% Cl: 0.05% - 0.75%) per year. The failure rate may increase in case of the Mireffik expulsion or perforation.
Levonorgestrel IUS may be particularly useful for contraception in patients with heavy menstrual bleeding, and can be successfully used in the treatment of idiopathic menorrhagia.
The volume of menstrual bleeding was decreased by 88% in menorrhagic women by the end of three months of use. Menorrhagia caused by submucosal fibroids may respond less favourably. Reduced bleeding promotes the increase of blood haemoglobin in patients with menorrhagia.
In idiopathic menorrhagia, prevention of proliferation of the endometrium is the probable mechanism of action of levonorgestrel IUS in reducing blood loss.
Bleeding Patterns:
Different kinds of bleeding changes (frequent, prolonged or heavy bleeding, spotting, oligomenorrhea, amenorrhoea) are experienced by all users of levonorgestrel IUS. In fertile women the average number of spotting days/month decreases gradually from nine to four days during the first six months of use. The percentage of women with prolonged bleeding (more than eight days) decreases from 20% to 3% during the first three months of use. In clinical studies during the first year of use, 17% of women experienced amenorrhoea of at least three months duration.
5.2 Pharmacokinetic properties
The initial release of levonorgestrel from Mireffik is 20 micrograms/24 hours, delivered directly into the uterine cavity. Because of the low plasma concentrations, there are only minor effects on the metabolism.
The pharmacokinetics of levonorgestrel itself have been extensively investigated and reported in the literature. A half life of 20 hours is considered the best estimate although some studies have reported values as short as 9 hours and others as long as 80 hours. Another important finding, although one in agreement with experience with other synthetic steroids, has been marked differences in metabolic clearance rates among individuals, even when administration was by the intravenous route. Levonorgestrel is extensively bound to proteins (mainly sex hormone binding globulin [SHBG]) and extensively metabolised to a large number of inactive metabolites.
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans other than the information already included in other sections of the SmPC. These data are based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential and toxicity to reproduction and development.
6 PHARMACEUTICAL PARTICULARS
6.1 List of excipients
Silicone base
Tetra-n-propyl silicate
Stannous octoate
Polydimethylsiloxane elastomer
Polydimethylsiloxane tubing
Polyethylene T-frame with 20-24% barium sulphate
Polypropylene thread
Copper phthalocyanine blue
6.2 Incompatibilities
Not applicable
6.3 Shelf life
48 months
6.4 Special precautions for storage
Store in the original package in order to protect from light.
6.5 Nature and contents of container
Mireffik IUS is individually packaged into a peel pouch bag. The peel pouches are made of 2 sheets:
1. PerfecFlex (clear Polyester-based Non-forming film with CPT which is a sealant film)
2. Tyvek 1073B (polyetylene fiber)
6.6 Special precautions for disposal
As the insertion technique is different from intrauterine devices, special emphasis should be given to training in the correct insertion technique. Special instructions for insertion are in the package.
Mireffik is supplied in a sterile pack which should not be opened until required for insertion. Each system should be handled with aseptic precautions. If the seal of the sterile envelope is broken, the system inside should be disposed of in accordance with the local guidelines for the handling of biohazardous waste. Likewise, a removed Mireffik and inserter should be disposed of in this manner. The outer carton package and the inner blister package can be handled as household waste.
7 MARKETING AUTHORISATION HOLDER
Mithra Pharmaceuticals S.A.
Rue Saint Georges, 5 4000 Liege Belgium
infomed.@mithra..be
8 MARKETING AUTHORISATION NUMBER(S)
PL 34096/0008
9 DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
06/08/2014
10 DATE OF REVISION OF THE TEXT
08/08/2016