Noqdirna 50 Microgram Oral Lyophilisate
SUMMARY OF PRODUCT CHARACTERISTICS
1 NAME OF THE MEDICINAL PRODUCT
Noqdirna 50 microgram oral lyophilisate
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
Each oral lyophilisate contains desmopressin acetate equivalent to 50 micrograms desmopressin.
For the full list of excipients, see section 6.1.
3 PHARMACEUTICAL FORM
Oral lyophilisate.
White, round, oral lyophilisate of approximately 12 mm marked with 50 on one side.
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
Noqdirna is indicated for symptomatic treatment of nocturia due to idiopathic nocturnal polyuria in adults (see section 5.1).
4.2 Posology and method of administration
Posology
• Women: 25 microgram daily, one hour before bedtime, administered sublingually without water.
• Men: 50 microgram daily, one hour before bedtime, administered sublingually without water.
A dose increase with this product is not recommended in elderly patients > 65 years.
If higher doses are considered for patients under the age of 65 years in case of an insufficient response to Noqdirna, other desmopressin oral lyophilisate products should be used (see sections 4.4, 4.8 and 5.1)
In the event of signs or symptoms of water retention and/or hyponatremia (headache, nausea/vomiting, weight gain, and, in severe cases, convulsions) treatment should be interrupted and reassessed. When restarting treatment strict fluid restriction should be enforced and serum sodium levels monitored (see section 4.4).
Noqdirna should be discontinued if the serum sodium level falls below the lower limit of normal range (i.e.135 mmol/L)
Special Populations
Elderly patients (65 years of age and older)
Elderly patients are at increased risk of developing hyponatraemia with desmopressin treatment and may also have impaired renal function. Caution should therefore be exercised in this age group and daily doses above 25 microgram for females and 50 microgram for males should not be used. In elderly patients serum sodium must be within the normal range, before initiating treatment, in the first week (4-8 days after initiation) and again at one month. Noqdirna should be discontinued if the serum sodium level falls below the lower limit of normal range (see section 4.4). Continued therapy must be carefully reconsidered in elderly patients who show no evidence of therapeutic benefit beyond 3 months.
Renal impairment
Noqdirna is contraindicated in patients with moderate and severe renal insufficiency (see section 4.3).
Hepatic impairment
No dose adjustment is needed for patients with hepatic impairment (see section 5.2).
Paediatric population
There is no relevant use of Noqdirna in the paediatric population for the indication of symptomatic treatment of nocturia due to idiopathic nocturnal polyuria.
Method of administration
Noqdirna is placed under the tongue where it dissolves without the need for water.
Food intake may reduce the intensity and duration of the antidiuretic effect at low doses of desmopressin (see section 5.2)
4.3 Contraindications
• Hypersensitivity to the active substances or to any of the excipients listed in section 6.1
• Habitual or psychogenic polydipsia (resulting in a urine production exceeding 40 ml/kg/24 hours)
• Known or suspected cardiac insufficiency or other conditions associated with fluid overload, sufficient to require treatment with diuretics, including a history of such conditions
• Moderate and severe renal insufficiency (creatinine clearance below 50 ml/min)
• Known history of hyponatremia
• Syndrome of inappropriate ADH secretion (SIADH)
4.4 Special warnings and precautions for use
Patients, in particular the elderly, should undergo clinical examination and questioning before commencing treatment with Noqdirna, given that nocturnal polyuria can be a symptom of cardiovascular or other medical conditions associated with fluid overload. If there is any suspicion of such coexistent conditions, treatment with desmopressin is not recommended (see also section 4.3).
Fluid intake must be limited to a minimum from 1 hour before until 8 hours after administration. Treatment without concomitant reduction of fluid intake may lead to prolonged fluid retention and/or hyponatremia with or without accompanying warning signs and symptoms (headache, nausea/vomiting, weight gain, and, in severe cases, convulsions).
Elderly patients with serum sodium levels in the lower range of normal may have an increased risk of hyponatremia. Patients 65 years and older should have their serum sodium monitored before initiating the treatment, in the first week of treatment (4-8 days) and again at one month after treatment initiation (see section 4.2).
At a 50 microgram dose level females may have an increased risk of hyponatraemia compared with males (see Section 5.1). It is therefore important that the gender-specific recommendations for dose are adhered to.
Noqdirna should be discontinued if the serum sodium level falls below the lower limit of normal range.
Desmopressin should be used with caution in patients with conditions characterized by fluid and/or electrolyte imbalance.
Treatment with desmopressin should be interrupted and reassessed during acute intercurrent illnesses characterised by fluid and/or electrolyte imbalance (such as systemic infections, fever, and gastroenteritis).
Precautions to avoid hyponatremia including careful attention to fluid restriction and more frequent monitoring of serum sodium must be taken in case of concomitant treatment with drugs, which are known to induce SIADH, e.g. tricyclic antidepressants, selective serotonin reuptake inhibitors, chlorpromazine, diuretics and carbamazepine, and some antidiabetics of the sulfonylurea group, particularly chlorpropamide, and in case of concomitant treatment with non-steroidal anti-inflammatory drugs (NSAIDs).
Special caution should be exercised in patients taking thiazide or loop diuretics for hypertension or other medical conditions not associated with fluid overload. Sodium monitoring in these patients is warranted.
Severe bladder dysfunction and outlet obstruction should be considered before starting treatment.
Caution is required in cases of cystic fibrosis, coronary heart disease, hypertensions, chronic renal disease and pre-eclampsia.
A diagnosis of nephrogenic diabetes insipidus should be considered if there is no reduction in night-time urine output after commencement of desmopressin.
Special caution should be exercised in patients taking lithium in case of masking of early-stage lithium-induced nephrogenic diabetes insipidus by administration of desmopressin for a nocturia indication. Desmopressin is not recommended in patients suspected of having lithium-induced nephrogenic diabetes insipidus.
4.5 Interaction with other medicinal products and other forms of interaction
Pharmacodynamic interactions
Substances, which are known to induce SIADH, may cause an increased risk of water retention/hyponatremia (e.g. tricyclic antidepressants, selective serotonin reuptake inhibitors, chlorpromazine, diuretics and carbamazepine as well as some antidiabetics of the sulfonylurea group particularly chlorpropamide) (see section 4.4).
NSAIDs and oxytocin may potentiate the antidiuretic effect of desmopressin and may induce water retention/ hyponatremia (see section 4.4).
Lithium may diminish the antidiuretic effect.
Pharmacokinetic interactions
Concomitant treatment with loperamide may result in a 3-fold increase of desmopressin plasma concentrations following oral administration, which may lead to an increased risk of water retention/hyponatremia. Although not investigated, other drugs slowing intestinal transport might have the same effect.
A standardised 27% fat meal significantly decreased absorption (rate and extent) of desmopressin tablets. No significant effect was observed with respect to pharmacodynamics (urine production or osmolality).
Food intake may reduce the intensity and duration of the antidiuretic effect at low oral doses of desmopressin tablet.
4.6 Fertility, pregnancy and lactation
Pregnancy
Caution should be exercised when prescribing to pregnant women.
Data on a limited number (n = 53) of exposed pregnancies in women with diabetes insipidus as well as data on a limited number of exposed pregnancies in women with bleeding complications (n=216) indicate no adverse effects of desmopressin on pregnancy or on the health of the foetus/new-born child. To date, no other relevant epidemiological data are available. Animal studies do not indicate direct or indirect harmful effects with respect to pregnancy, embryonic/foetal development, parturition or postnatal development.
Animal reproduction studies have shown no clinically relevant effects on parents and offspring. In-vitro analysis of human cotyledon models have shown that there is no transplacental transport of desmopressin when administered at therapeutic concentration corresponding to recommended dose.
Breastfeeding
Results from analyses of milk from nursing mothers receiving high dose desmopressin acetate (300 microgram intranasal); indicate that the amounts of desmopressin that may be transferred to the child are considerably less than the amounts required to influence diuresis. Therefore it is not considered necessary to stop breastfeeding.
Fertility
Studies with desmopressin in animals have shown no impairment of fertility in male and female rats.
4.7 Effects on ability to drive and use machines
Noqdirna has no or negligible influence on the ability to drive and use machines.
4.8 Undesirable effects
Summary of the safety profile
Based on the frequency of adverse drug reactions reported in clinical studies with Noqdirna for nocturia indication conducted in male subjects (50 mcg; N=222) and in female subjects (25 mcg; N=219) the most commonly reported adverse reaction during treatment was dry mouth (13%), headache (3%), hyponatraemia (3%), and dizziness (2%).
Description of selected adverse reactions:
The most serious adverse reaction with desmopressin is hyponatraemia, which is associated with headache, nausea, vomiting, decreased serum sodium, weight increase, malaise, abdominal pain, muscle cramps, dizziness, confusion, decreased consciousness and in severe cases convulsions and coma. The hyponatraemia is an antidiuretic effect, arising from increased water reabsorption by the renal tubules and osmotic dilution of plasma. In studies with adult subjects treated for nocturia, the majority of the subjects developed low serum sodium within the first days of treatment or in relation to dose increase. Special attention should be paid to the precautions addressed in section 4.4.
Females have a higher risk of hyponatraemia which may be due to increased sensitivity of the kidney tubules to vasopressin and its analogues in women compared with men. The risk of this is minimised by recommendation of a lower dose in women. The risk of hyponatraemia in the over 65 years age group is further reduced by monitoring of serum sodium in this age group (see section 4.2 and 4.4).
Tabulated list of adverse reactions
The below table 1 shows the frequencies of adverse reactions reported. The frequencies are defined as follows: very common (>1/10), common (>1/100 to <1/10) and uncommon (>1/1,000 to <1/100).
Table 1: Frequency of adverse drug reactions reported (Phase III studies and Post-marketing reports)
|
MedDRA System Organ Class |
Very common (> 1/10) |
Common (> 1/100 to < 1/10) |
Uncommon (> 1/1,000 to < 1/100) |
|
Metabolism and nutrition disorders |
Hyponatraemia | ||
|
Nervous system disorders |
Headache Dizziness | ||
|
Gastrointestinal disorders |
Dry mouth* |
Nausea Diarrhoea |
Constipation Abdominal discomfort |
|
General disorders and administration site conditions |
Fatigue Oedema peripheral |
*It is to be noted that subjects were specifically queried about dry mouth in some of the clinical studies.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Yellow Card Scheme, website: www.mhra.gov.uk/yellowcard.
4.9 Overdose
Symptoms:
Overdose of Noqdirna leads to a prolonged duration of action with an increased risk of water retention and hyponatremia.
Treatment:
Although the treatment of hyponatraemia should be individualised, the following general recommendations can be given. Hyponatraemia is treated by discontinuing the desmopressin treatment, fluid restriction and symptomatic treatment if needed.
5 PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Vasopressin and analogues. ATC code: H01B A02
Mechanism of action
Noqdirna contains desmopressin a synthetic analogue of naturally occurring anti-diuretic hormone arginine vasopressin (AVP). Desmopressin mimics vasopressin’s anti-diuretic effect, binding to the V2 receptors in the renal collecting tubules of the kidneys, causing reabsorption of water into the body. This reabsorption in turn decreases night-time urine production. Due to the proposed low gender-specific doses (25 microgram for females and 50 microgram for males), and the limited duration of action of Noqdirna, the antidiuretic activity is limited to the night-time sleep period.
Pharmacodynamic effects
In study CS29, the weight-corrected Noqdirna dose that induced 50% maximum achievable drug effect on nocturnal urine volume differed significantly between females and males. The estimated exposure value for males was 2.7-fold (95% CI: 1.3-8.1) higher than the value for females to obtain an identical dynamic effect, corresponding to higher desmopressin sensitivity among females. The development of hyponatremia is dose dependent. Females are at higher risk of developing hyponatraemia than males. The incidences of hyponatremia rises with increasing age (see section
4.2 and 4.4).
Clinical efficacy
The efficacy of Noqdirna has been demonstrated in two randomised double blinded placebo controlled studies in respectively 268 women (study CS40, desmopressin Melt 25 microgram versus placebo) and 395 men (study CS41, desmopressin Melt 50 microgram and 75 microgram versus placebo) with nocturia defined as an average of >2 nocturnal voids per night and polyuria in 90% of women and 87% of men.
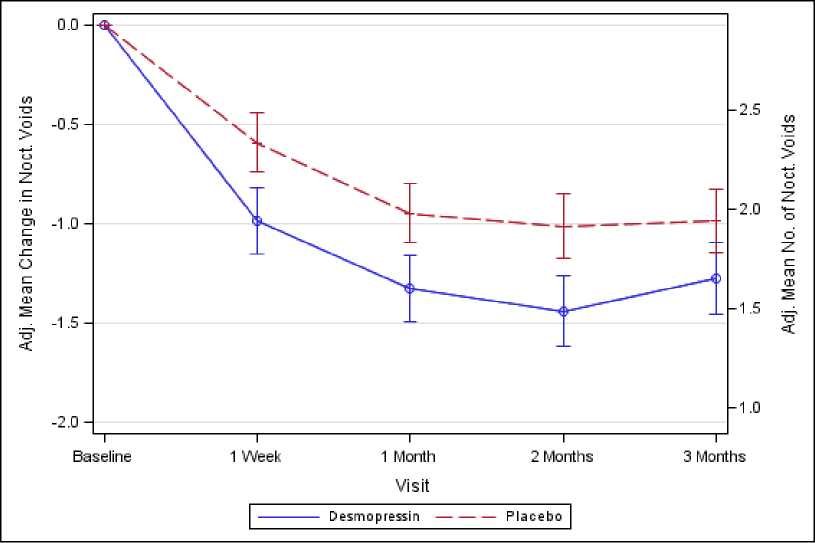
Both studies met the 2 co-primary endpoints with statistically significant differences favouring desmopressin Melt over the 3-month period. There was a statistically significant decrease in the adjusted mean number of nocturnal voids from the baseline on desmopressin Melt 25 microgram (-1.46) compared to placebo (-1.24) in the female study (p=0.028) (Fig. 1) and on desmopressin Melt 50 microgram (-1.25) compared to placebo (-0.88) in the male study (p=0.0003) (Fig. 2). The proportion of subjects with >33% decrease in the mean number of nocturnal voids (responders) was significantly increased, nearly doubled. The odds ratio for >33% decrease of desmopressin Melt 25 microgram compared to placebo was 1.85 (p=0.006) in the female study and
the odds ratio for >33% decrease of desmopressin Melt 50 microgram compared to placebo was 1.98 (p=0.0009) in the male study.
For secondary endpoints, there was an increase from baseline to 3 months in the first undisturbed sleep period (FUSP)/time to first void with a treatment contrast of 49 minutes in the female study and 39 minutes in the male study. There was a statistically significant improvement in quality of life for desmopressin Melt 25 microgram (N-QoL total score 27.24) compared to placebo (21.90) (p=0.0226) in female and an improvement for desmopressin Melt 50 microgram (N-QoL total score 18.37) compared to placebo (13.88) (p=0.0385) in male. There was a strong association (p<0.0001) in the both studies between treatment response (reduction in number of nocturnal voids and increase in FUSP) and improvements in patients’ quality of life.
Figure 1. Co-Primary Endpoint: Adjusted mean change from baseline in nocturnal voids during 3 months of treatment - (Females, CS40 Full Analysis Set)
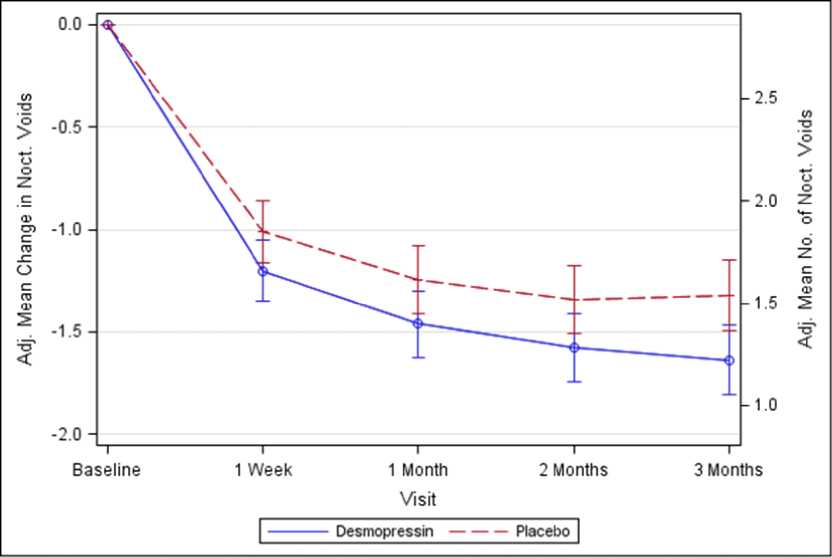
Figure 2. Co-Primary Endpoint: Adjusted mean change from baseline in nocturnal voids during 3 months of treatment - (Males, CS41 Full Analysis Set)
In a double-blind randomised clinical study, the efficacy and safety of a combination therapy with desmopressin Melt and tolterodine extended release capsules was investigated for the treatment of overactive bladder with nocturia in women, for a period of 3 months. Forty-nine subjects were exposed to a combination of Noqdirna (desmopressin Melt) 25 microgram and tolterodine 4 milligram. No serious adverse events were observed in this study and safety profile of the combination treatment was similar to the safety profile of Noqdirna 25 microgram. The efficacy in terms of reduction from baseline in mean number of nocturnal voids during 3 months treatment was numerically greater in the combination therapy group versus tolterodine monotherapy group (treatment contrast, -0.34 voids) in full analysis set, and the difference reached statistical significance (p=0.049) with a treatment contrast of -0.41 voids in the per protocol analysis set.
Gender differences in clinical safety and efficacy
Clinical study [FE992026 CS029] analysed the dose-response to Noqdirna in females and males at doses ranging from 10 to 100 microgram: In females, there was no further gain in pharmacodynamic effect above the dose of 25 microgram, indicating that the dose response plateau was reached at 25 microgram in females. In males, reduction in urine volume was greater at 50 microgram, but not substantially higher at 100 microgram. Increasing doses to 50 microgram dose level in females did not yield further efficacy, but was associated with a 5-fold increase in the risk of hyponatraemia compared with males in the age group above 50 years (p = 0.015).
5.2 Pharmacokinetic properties
Absorption
The overall mean absolute bioavailability of desmopressin administered sublingually from earlier dose-ranging studies of doses of 200, 400 and 800 mcg is 0.25%, with a 95% confidence interval of 0.21 - 0.31%. Desmopressin exhibits a moderate-to-high variability in bioavailability, both within and between subjects. Desmopressin shows dose linearity regarding AUC and Cmax in the range of 60 to 240 mcg. However, the bioavailability of doses below 60 has not been evaluated.
Distribution
The distribution of desmopressin is best described by a two-compartment distribution model with a volume of distribution during the elimination phase of 0.3-0.5 L/kg.
Biotransformation
The in-vivo metabolism of desmopressin has not been studied. In vitro human liver microsome metabolism studies of desmopressin have shown that no significant amount is metabolized in the liver by the cytochrome P450 system. Thus human liver metabolism in vivo by the cytochrome P450 system is unlikely to occur. The effect of desmopressin on the PK of other drugs is likely to be minimal due to its lack of inhibition of the cytochrome P450 drug metabolizing system.
Elimination
The total clearance of desmopressin has been calculated to 7.6 L/hr. The terminal half-live of desmopressin is estimated to 2.8 hours. In healthy subjects the fraction excreted unchanged was 52 % (44 % - 60 %).
Linearity/non-linearity
There are no indications of non-linearities in any of the pharmacokinetic parameters of desmopressin.
Characteristics in specific groups of patients
Renal impairment:
Depending on the degree of renal impairment the AUC and half-live increased with the severity of the renal impairment. Desmopressin is contraindicated in patients with moderate and severe renal impairment (creatinine clearance below 50 ml/min).
Table 2: Pharmacokinetic parameters for different degrees of renal impairment. Data from CS001.
|
Creatinine Clearance |
Renal Function |
AUC (Hrs*pg/mL) |
Ty2 (Hrs) | |
|
Healthy |
>80 mL/min |
Normal |
186 |
2.8 |
|
Mild |
50-80 mL/min |
Mildly impaired |
281 |
4.0 |
|
Moderate |
30-49 mL/min |
Moderately impaired |
453 |
6.7 |
|
Severe |
5-29 mL/min |
Severely impaired |
682 |
8.7 |
Hepatic impairment:
No studies have been performed in this population.
It is unlikely that desmopressin will interact with drugs affecting hepatic metabolism, since desmopressin has been shown not to undergo significant liver metabolism in in vitro studies with human microsomes.
5.3 Preclinical safety data
Non-clinical data revealed no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity and toxicity to reproduction.
Carcinogenicity studies have not been performed with desmopressin, because it is closely related to the naturally-occurring peptide hormone.
6 PHARMACEUTICAL PARTICULARS
6.1 List of excipients
Gelatin
Mannitol (E 421) Citric acid, anhydrous
6.2 Incompatibilities
Not applicable.
Shelf life
6.3
4 years
6.4 Special precautions for storage
This medicinal product does not require any special temperature storage conditions.
Store in the original package in order to protect from moisture and light.
Use immediately upon opening individual tablet blister
6.5 Nature and contents of container
Perforated unit dose blisters packed in a carton. The blister bottom foil and the blister lid foil are multilayer laminates consisting of PVC/OPA/Alu/OPA/PVC and heat seal laquer/Alu/PET/paper, respectively.
Pack size:
10x1, 30x1, 90x1 or 100x1 oral lyophilisates Not all pack sizes may be marketed.
6.6 Special precautions for disposal
No special requirements.
Any unused medicinal product or waste material should be disposed in accordance with local requirements.
7 MARKETING AUTHORISATION HOLDER
Ferring Pharmaceuticals Ltd
Drayton Hall Church Road West Drayton UB7 7PS
8 MARKETING AUTHORISATION NUMBER(S)
PL 03194/0119
9 DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
18/05/2016
10 DATE OF REVISION OF THE TEXT
18/05/2016