Prenotrix 35 Microgram /H Transdermal Patch
SUMMARY OF PRODUCT CHARACTERISTICS
1 NAME OF THE MEDICINAL PRODUCT
Prenotrix 35 microgram /h transdermal patch
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
Prenotrix 35 micrograms/h
Each transdermal patch contains 20 mg buprenorphine.
Area containing the active substance: 25 cm2
Nominal release rate: 35 micrograms of buprenorphine per hour.
Excipient: soya oil 16 mg
For a full list of excipients, see section 6.1.
3 PHARMACEUTICAL FORM
Transdermal patch
The patches are tan coloured, rectangular with four rounded edges and topped off corners and labelled with Buprenorphin 35 pg/h.
Each patch is packed in single sealed sachets.
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
Moderate to severe cancer pain and severe pain which does not respond to non-opioid analgesics.
Prenotrix is not suitable for the treatment of acute pain.
4.2 Posology and method of administration
Patients over 18 years of age
The Prenotrix dosage should be adapted to the condition of the individual patient (pain intensity, suffering, individual reaction). The lowest possible dosage providing adequate pain relief should be given. Three transdermal patch strengths are available to provide such adaptive treatment: Prenotrix 35 micrograms/h, Prenotrix 52.5 micrograms/h and Prenotrix 70 micrograms/h.
Initial dose selection:
Patients who have previously not received any analgesics should start with the lowest transdermal patch strength (Prenotrix 35 micrograms/h). Patients previously given a WHO step-I analgesic (non-opioid) or a step-II analgesic (weak opioid) should also begin with Prenotrix 35 micrograms/h. According to the WHO recommendations, the administration of a non-opioid analgesic can be continued, depending on the patient's overall medical condition.
When switching from a step-III analgesic (strong opioid) to Prenotrix and choosing the initial transdermal patch strength, the nature of the previous medication, administration and the mean daily dose should be taken into account in order to avoid the recurrence of pain. In general it is advisable to titrate the dose individually, starting with the lowest transdermal patch strength (Prenotrix 35 micrograms/h). Clinical experience has shown that patients who were previously treated with higher daily dosages of a strong opioid (in the dimension of approximately 120 mg oral morphine) may start the therapy with the next higher transdermal patch strength (see also section 5.1).
To allow for individual dose adaptation in an adequate time period sufficient supplementary immediate release analgesics should be made available during dose titration.
The necessary strength of Prenotrix must be adapted to the requirements of the individual patient and checked at regular intervals.
After application of the first Prenotrix transdermal patch the buprenorphine serum concentrations rise slowly both in patients who have been treated previously with analgesics and in those who have not. Therefore initially, there is unlikely to be a rapid onset of effect. Consequently, a first evaluation of the analgesic effect should only be made after 24 hours.
The previous analgesic medication (with the exception of transdermal opioids) should be given in the same dose during the first 12 hours after switching to Prenotrix and appropriate rescue medication on demand in the following 12 hours.
Dose titration and maintenance therapy Prenotrix can be applied for maximal 72 hours.
Prenotrix should be replaced after 72 hours (3 days) at the latest. The dose should be titrated individually until analgesic efficacy is attained. If analgesia is insufficient at the end of the initial application period, the dose may be increased, either by applying more than one transdermal patch of the same strength or by switching to the next transdermal patch strength. At the same time no more than two transdermal patches regardless of the strength should be applied.
Before application of the next Prenotrix strength the amount of total opioids administered in addition to the previous transdermal patch should be taken into consideration, i.e. the total amount of opioids required, and the dosage adjusted accordingly. Patients requiring a supplementary analgesic (e.g. for breakthrough pain) during maintenance therapy may take for example one to two 0.2 mg buprenorphine sublingual buprenorphine every 24 hours in addition to the transdermal patch. If the regular addition of 0.4 - 0.6 mg sublingual buprenorphine is necessary, the next strength should be used.
Patients under 18 years of age
As Prenotrix has not been studied in patients under 18 years of age, the use of the medicinal product in patients below this age is not recommended.
Elderly patients
No dosage adjustment of Prenotrix is required for elderly patients.
Patients with renal insufficiency
Since the pharmacokinetics of buprenorphine is not altered during the course of renal failure, its use in patients with renal insufficiency, including dialysis patients, is possible.
Patients with hepatic insufficiency
Buprenorphine is metabolised in the liver. The intensity and duration of its action may be affected in patients with impaired liver function. Therefore patients with liver insufficiency should be carefully monitored during treatment with Prenotrix.
Method of administration
Prenotrix should be applied to non-irritated, clean skin on a non-hairy flat surface, but not to any parts of the skin with large scars. Preferable sites on the upper body are: upper back or below the collar-bone on the chest. Any remaining hairs should be cut off with a pair of scissors (not shaved). If the site of application requires cleansing, this should be done with water. Soap or any other cleansing agents should not be used. Skin preparations that might affect adhesion of the transdermal patch to the area selected for application of Prenotrix should be avoided.
The skin must be completely dry before application. Prenotrix is to be applied immediately after removal from the sachet.
Prenotrix should be worn continuously for up to 3 days. After removal of the previous transdermal patch a new Prenotrix transdermal patch should be applied to a different skin site. At least one week should elapse before a new transdermal patch is applied to the same area of skin.
How to apply the patch
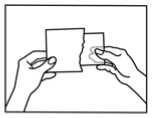
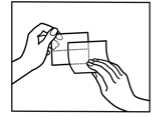
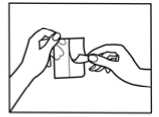
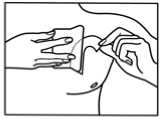
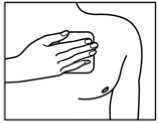
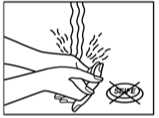
1. The sachet must not be opened, before actually intending to use the patch.
2. At first the loose separation foil has to be removed.
3. Afterwards one half of the strip-foil of the patch should be removed but without touching the sticking part.
4. The other half of the strip-foil can be removed after applying one half of the patch onto the skin.
5. The patch has to be pressed firmly onto the skin with the palm of the hand for 30 to 60 seconds to ensure that the patch is well stuck to the skin, especially around the edges.
6. After using the patch the hands must be washed without any cleansing products.
Provided the patch is applied correctly, it is very unlikely to come unstuck. Taking a shower, a bath or swimming while wearing the patch is possible but any external heat exposure of the patch (i.e. sauna, infra-red radiation) should be avoided.
However if the patch starts to come off prior to the next change, the same patch cannot be used again but a new patch has to be applied immediately.
How to change the patch
• Removal of the old patch
• Both sticking ends of the patch must be folded together the sticking parts being on the inside.
• The patch must be carefully disposed.
• A new patch should be applied to another suitable skin area (as detailed above). The same skin area can only be used again after 2 applications.
Duration of treatment
Prenotrix should under no circumstances be administered for longer than absolutely necessary. If long-term treatment with Prenotrix appears necessary in view of the nature and severity of the illness, then careful and regular monitoring should be carried out (if necessary with breaks in treatment) to establish whether and to what extent further treatment is necessary.
Discontinuation of buprenorphine patch
After removal of Prenotrix buprenorphine serum concentrations decrease gradually and thus the analgesic effect is maintained for a certain amount of time. This should be considered when therapy with Prenotrix is to be followed by other opioids. As a general rule, a subsequent opioid should not be administered within 24 hours after removal of Prenotrix. For the time being only limited information is available on the starting dose of other opioids administered after discontinuation of Prenotrix.
4.3 Contraindications
Prenotrix is contraindicated in:
- hypersensitivity to the active substance buprenorphine, soya, peanuts or to any of the excipients (for the excipients, see section 6.1)
- in opioid-dependent patients and for narcotic withdrawal treatment
- conditions in which the respiratory centre and function are severely impaired or may become so
- patients who are receiving MAO inhibitors or have taken them within the last two weeks (see section 4.5)
- patients suffering from myasthenia gravis patients suffering from delirium tremens
4.4 Special warnings and precautions for use
Prenotrix must only be used with particular caution in acute alcohol intoxication, convulsive disorders, in patients with head injury, shock, a reduced level of consciousness of uncertain origin, increased intracranial pressure without the possibility of ventilation.
Buprenorphine occasionally causes a respiratory depression. Therefore care should be taken when treating patients with impaired respiratory function or patients receiving medicinal products which can cause respiratory depression.
Buprenorphine has a substantially lower dependence liability than pure opioid agonists. In healthy volunteer and patient studies with Prenotrix, withdrawal reactions have not been observed. However, after long-term use of Prenotrix withdrawal symptoms, similar to those occuring during opiate withdrawal, cannot be entirely excluded (see section 4.8). These symptoms are: agitation, anxiety, nervousness, insomnia, hyperkinesia, tremor and gastro-intestinal disorders.
In patients abusing opioids, substitution with buprenorphine may prevent withdrawal symptoms. This has resulted in some abuse of buprenorphine and caution should be exercised when prescribing it to patients suspected of having drug abuse problems.
Buprenorphine is metabolised in the liver. The intensity and duration of effect may be altered in patients with liver function disorders. Therefore such patients should be carefully monitored during Prenotrix treatment.
Buprehnorphin is listed from the World Anti-Doping Agency as substance prohibited in competition.
Fever / External use of heat
Fever and the presence of heat may increase the permeability of the skin. Theoretically in such situations buprenorphine serum concentrations may be raised during Prenotrix treatment. Therefore on treatment with Prenotrix, attention should be paid to the increased possibility of opioid reactions in febrile patients or those with increased skin temperature due to other causes.
4.5 Interaction with other medicinal products and other forms of interaction
On administration of MAO-inhibitors in the last 14 days prior to the administration of the opioid pethidine threatening interactions have been observed affecting the central nervous system and respiratory and cardiovascular function. The same interactions of MAO-inhibitors and Prenotrix cannot be ruled out (see section 4.3).
When Prenotrix is applied together with other opioids, anaesthetics, hypnotics, sedatives, antidepressants, neuroleptics and in general medicinal products that depress respiration and the central nervous system, the CNS effects may be intensified. This applies also to alcohol.
Administered together with inhibitors or inducers of CYP 3A4 the efficacy of Prenotrix may be intensified (inhibitors e.g. ketoconazole) or weakened (inducers e.g. phenobarbital, carbamazepine, phenytoin and rifampicin).
4.6 Fertility, pregnancy and lactation
Pregnancy
There are limited amount of data from the use of Prenotrix in pregnant women; studies in animals have shown reproductive toxicity (see section 5.3). The potential risk for humans is unknown.
Towards the end of pregnancy high doses of buprenorphine may induce respiratory depression in the new-born infants even after a short period of administration.
Long-term administration of buprenorphine during the last three months of pregnancy may cause a withdrawal syndrome in the new born infants.
Therefore Prenotrix should not be used during pregnancy and in women of childbearing potential who are not using effective contraception.
Lactation
Buprenorphine is excreted in human milk. In rats, buprenorphine has been found to inhibit lactation.
Prenotrix should not be used during lactation.
4.7 Effects on ability to drive and use machines
Even when used according to instructions, this medicine can impair cognitive function and can affect a patient’s ability to drive and use machines safely. This applies particularly in conjunction with other centrally acting substances including alcohol, tranquilizer, sedatives and hypnotics.
This class of medicine is in the list of drugs included in regulations under 5a of the Road Traffic Act 1988. When prescribing this medicine, patients should be told:
• The medicine is likely to affect your ability to drive
• Do not drive until you know how the medicine affects you
• It is an offence to drive while under the influence of this medicine
• However, you would not be committing an offence (called ‘statutory defence’) if:
o the medicine has been prescribed to treat a medical or dental problem and;
o you have taken it according to the instructions given by the prescriber and in the information provided with the medicine and;
o it was not affecting your ability to drive safely
Patients wearing a Prenotrix transdermal patch should not drive or use machines, nor for at least 24 hours after the transdermal patch has been removed.
4.8 Undesirable effects
The following adverse reactions were reported after administration of buprenorphine in clinical studies and from post marketing surveillance.
The frequencies are given as follows:
Very common (>1/10)
Common (>1/100 to <1/10)
Uncommon (>1/1.000 to <1/100)
Rare (>1/10.000 to <1/1.000)
Very rare (<1/10.000)
Not known (cannot be estimated from the available data)
The most commonly reported systemic adverse reactions were nausea and vomiting.
The most commonly local adverse reactions were erythema and pruritus.
Immune system disorders
Very rare: serious allergic reactions
Metabolism and nutrition disorders
Rare: appetite lost
Psychiatric disorders
Uncommon: confusion, sleep disorder, restlessness
Rare: psychotomimetic effects (e.g. hallucinations, anxiety, nightmares), decreased libido
Very rare: dependence, mood swings
Nervous system disorders
Common: headache, dizziness
Uncommon: Sedation, somnolence
Rare: concentration impaired, speech disorder, numbness, dysequilibrium, paraesthesia (e.g. pricking or burning skin sensation)
Very rare: muscle fasciculation, parageusia
Eye disorders
Rare: visual disturbance, blurring of vision, eyelid oedema Very rare: miosis
Ear and labyrinth disorders
Very rare: ear pain
Cardiac/Vascular disorders
Uncommon: circulatory disorders (such as hypotension or, rarely, even circulatory collapse)
Rare: hot flushes
Respiratory, thoracic and mediastinal disorders
Common: dyspnoea
Rare: respiratory depression
Very rare: hyperventilation, hiccups
Gastrointestinal disorders
Very common: nausea
Common: vomiting, constipation
Uncommon: dry mouth Rare: pyrosis Very rare: retching
Skin and subcutaneous tissue disorders
Very common: erythema, pruritus
Common: exanthema, diaphoresis
Uncommon: rash Rare: urticaria
Very rare: pustules, vesicles
Renal and urinary disorders
Uncommon: urinary retention, micturition disorders
Reproductive system and breast disorders
Rare: decreased erection
General disorders and administration site conditions
Common: oedema, tiredness
Uncommon: weariness
Rare: withdrawal symptoms, administration site reactions Very rare: thoracic pain
In some cases delayed allergic reactions occurred with marked signs of inflammation. In such cases treatment with Prenotrix should be terminated.
Buprenorphine has a low risk of dependence. After discontinuation of Prenotrix, withdrawal symptoms are unlikely. This is due to the very slow dissociation of buprenorphine from the opiate receptors and to the gradual decrease of buprenorphine serum concentrations (usually over a period of 30 hours after removal of the last transdermal patch). However, after long-term use of Prenotrix withdrawal symptoms, similar to those occurring during opiate withdrawal, cannot be entirely excluded. These symptoms include: agitation, anxiety, nervousness, insomnia, hyperkinesia, tremor and gastrointestinal disorders.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via Yellow Card Scheme [www.mhra.gov.uk/yellowcard].
4.9 Overdose
Buprenorphine has a wide safety margin. Due to the rate-controlled delivery of small amounts of buprenorphine into the blood circulation high or toxic buprenorphine concentrations in the blood are unlikely. The maximum buprenorphine serum concentration after the application of the Prenotrix 70 micrograms/h patch is about six times less than after the intravenous administration of the therapeutic dose of 0.3 mg buprenorphine.
Symptoms
In principal, on overdose with buprenorphine, symptoms similar to those of other centrally acting analgesics (opioids) are to be expected. These are: respiratory depression, sedation, somnolence, nausea, vomiting, cardiovascular collapse, and marked miosis.
Treatment
General emergency measures apply. Keep the airway open (aspiration!). Maintain respiration and circulation depending on the symptoms. Naloxone has a limited impact on the respiratory depressant effect of buprenorphine. High doses are needed given either as repeated boluses or infusion (for example starting with a bolus administration of 1-2 mg intravenously. Having attained an adequate antagonistic effect, administration by infusion is recommended to maintain constant naloxone plasma levels). Therefore, adequate ventilation should be established.
5 PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: opioids, oripavine derivatives, ATC-Code: N02AE01.
Buprenorphine is a strong opioid with agonistic activity at the mu-opioid receptor and antagonistic activity at the kappa-opioid receptor. Buprenorphine appears to have the general characteristics of morphine, but has its own specific pharmacology and clinical attributes.
In addition, numerous factors, e.g. indication and clinical setting, route of administration and the interindividual variability, have an impact on analgesia and therefore have to be considered when comparing analgesics.
In daily clinical practice different opioids are ranked by a relative potency, although this is to be considered a simplification.
The relative potency of buprenorphine in different application forms and in different clinical settings has been prescribed in literature as follows:
• Morphine p.o. : BUP i.m. as 1 : 67 - 150 (single dose; acute pain model)
• Morphine p.o. : BUP s.l. as 1 : 60 - 100 (single dose, acute pain model; model dose, chronic pain, cancer pain)
• Morphine p.o. : BUP TTS as 1 : 75 - 115 (multiple dose, chronic pain)
Adverse reactions are similar to those of other strong opioid analgesics. Buprenorphine appears to have a lower dependence liability than morphine.
5.2 Pharmacokinetic properties
General characteristics of the active substance
Buprenorphine has a plasma protein binding of about 96 %.
Buprenorphine is metabolised in the liver to N-dealkylbuprenorphine (norbuprenorphine) and to glucuronide conjugated metabolites. 2/3 of the active substance is eliminated unchanged in the faeces and 1/3 eliminated as conjugates of unchanged or dealkylated buprenorphine via the urinary system. There is evidence of enterohepatic recirculation.
Studies in non-pregnant and pregnant rats have shown that buprenorphine passes blood-brain and placental barriers. Concentrations in the brain (which contained only unchanged buprenorphine) after parenteral administration were 2-3 times higher than after oral administration.
After intra-muscular or oral administration buprenorphine apparently accumulates in the foetal gastrointestinal-lumen - presumably due to biliary excretion, as enterohepatic circulation has not fully developed.
Properties of buprenorphine with healthy test persons
After the application of Prenotrix, buprenorphine is absorbed through the skin. The continuous delivery of buprenorphine into the systemic circulation is by controlled release from the adhesive polymer-matrix-system.
After the initial application of Prenotrix the plasma concentrations of buprenorphine gradually increase, and after 4 to 12 h the plasma concentrations reach the minimum effective concentration of 100 pg/ml. From the studies performed with Prenotrix 35 micrograms/h an average Cmax of 273 pg/ml and an average tmax of 34 h from studies performed with Prenotrix 70 micrograms/h an average Cmax of 425 pg/ml and an average tmax of 29 h were determined.
After removal of Prenotrix the plasma-concentrations of buprenorphine steadily decrease and are eliminated with a half-life of approx.25 hours (range 24-27). Due to the continuous absorption of buprenorphine from the depot in the skin elimination is than after intravenous administration.
5.3 Preclinical safety data
Standard toxicological studies have not shown evidence of any particular potential risks for humans. In tests with repeated doses of buprenorphine in rats the increase in body weight was reduced.
Studies on fertility and general reproductive capacity of rats showed no detrimental effects. Studies in rats and rabbits revealed signs of fetotoxicity and increased postimplantation loss.
Studies in rats showed diminished intra-uterine growth, delays in the development of certain neurological functions and high peri/postnatal mortality in the new-born infants after treatment of the dams during gestation or lactation.
There is evidence that complicated delivery and reduced lactation contributed to these effects. There was no evidence of embryotoxicity including teratogenicity in rats or rabbits.
In vitro and in vivo examinations on the mutagenic potential of buprenorphine did not indicate any clinically relevant effects.
In long-term studies in rats and mice there was no evidence of any carcinogenic potential relevant for humans.
Toxicological data available did not indicate a sensitising potential of the additives of the transdermal patches.
6 PHARMACEUTICAL PARTICULARS
6.1 List of excipients
Drug containing adhesive matrix:
styrene-butadiene-styrene (SBS) and styrene-butadiene block co-polymers, colophonium resin, antioxidants (2,4-Bis(1,1-Dimethylethyl)phenyl phosphite (3:1); Tris(2,4-Di-Tert-Butylphenyl)phosphate), aloe vera leaf extract oil (also contains refined soya-bean oil and alpha tocopherol acetate)
Backing foil: pigmented polyethylene and aluminium vapour coated polyester, blue printing colour
Release liner with pull of aid: polyester film, one side siliconised (to be removed prior application)
6.2 Incompatibilities
Not applicable
6.3 Shelf life
18 months.
6.4 Special precautions for storage
Do not store above 25°C
Do not freeze
6.5 Nature and contents of container
Each transdermal patch is covered with a loose siliconised PETP foil cover sheet and is packed individually into a sealed sachet. The sachet is made from PETP/Aluminium/PE. Packs contain 4, 5, 8, 10, 16, 20 or 24 (6 x 4) individually sealed patches.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal
Large quantities of buprenorphine remain in the transdermal patches even after use. Used transdermal patches should be folded with the adhesive surface inwards and discarded or whenever possible returned to the pharmacy. Any unused medicinal product should be discarded or returned to the pharmacy.
7 MARKETING AUTHORISATION HOLDER
Activase Pharmaceuticals Limited,
11 Boumpoulinas, P.C. 1060 Nicosia.
Cyprus
8 MARKETING AUTHORISATION NUMBER(S)
PL 28444/0130
9 DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
April 2013/ 18/02/2014
10 DATE OF REVISION OF THE TEXT
18/02/2014