Propinorm Xl 30 Mg Modified-Release Capsules
SUMMARY OF PRODUCT CHARACTERISTICS
1 NAME OF THE MEDICINAL PRODUCT
Propinorm XL 30 mg Modified-Release Capsules
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
Each capsule contains 30 mg propiverine hydrochloride (equivalent to 27.28 mg propiverine).
Excipients: Lactose monohydrate (5.7 mg), for a full list of excipients, see section 6.1
3 PHARMACEUTICAL FORM
Modified-release capsule, hard
Orange and white size 3 capsules containing white to off-white pellets.
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
Symptomatic treatment of urinary incontinence and/or increased urinary frequency and urgency as may occur in patients with overactive bladder.
4.2 Posology and method of administration
Capsules for oral use.
Do not crush or chew the capsules.
The recommended daily doses are as follows:
Adults: As a standard dose one capsule (= 30 mg propiverine hydrochloride) once a day is recommended.
Elderly: Generally there is no special dosage regimen for the elderly (see section 5.2). Paediatric population: Due to a lack of data, this product should not be used in children.
Caution should be exercised and clinicians should monitor patients carefully for side effects in the following dispositions (see sections 4.4, 4.5, 5.2) .
Use in renal impairment
In patients with mild to moderate impaired renal function there is no need for a dose adjustment (see section 5.2).
Use in hepatic impairment
In patients with mild impaired hepatic function there is no need for a dose adjustment but caution should be exercised. The treatment of patients with moderate to severe impairment is not recommended because no data are available (see section 5.2).
Patients receiving concomitant treatment with drugs that are potent inhibitors of CYP 3A4 combined with methimazole
In patients receiving drugs that are potent FMO inhibitors such as methimazole in combination with potent CYP 3A4/5 inhibitors treatment should start with a dose of 15 mg per day. The dose may be titrated to a higher dose. However, caution should be exercised and clinicians should monitor these patients carefully for side effects (see sections 4.4, 4.5, 5.2).
There is no clinically relevant effect of food on the pharmacokinetics of propiverine (see section 5.2). Accordingly, there is no particular recommendation for the intake of propiverine in relation to food.
4.3 Contraindications
The drug is contraindicated in patients who have demonstrated hypersensitivity to the active substance or to any of the excipients and in patients suffering from one of the following disorders:
obstruction of the bowel
significant degree of bladder outflow obstruction where urinary retention may be anticipated
myasthenia gravis
intestinal atony
severe ulcerative colitis
toxic megacolon
uncontrolled angle closure glaucoma moderate or severe hepatic impairment tachyarrhythmias
4.4 Special warnings and precautions for use
The drug should be used with caution in patients suffering from:
autonomic neuropathy severe renal impairment
Symptoms of the following diseases may be aggravated following administration of the drug:
severe congestive heart failure (NYHA IV) prostatic hypertrophy hiatus hernia with reflux oesophagitis cardiac arrhythmia tachycardia
Propiverine, like other anticholinergics, induces mydriasis. Therefore, the risk to induce acute angle-closure glaucoma in individuals predisposed with narrow angles of the anterior chamber may be increased. Drugs of this class have been reported to induce or precipitate acute angle-closure glaucoma.
Pollakiuria and nocturia due to renal disease or congestive heart failure as well as organic bladder diseases (e.g. urinary tract infections, malignancy) should be ruled out prior to treatment.
This product contains lactose monohydrate. Patients with rare hereditary problems of galactose intolerance, the lapp lactose deficiency or glucose-galactose malabsorption should not take this medication.
4.5 Interaction with other medicinal products and other forms of interaction
Increased effects due to concomitant medication with tricyclic antidepressants (e. g. imipramine), tranquillisers (e.g. benzodiazepines), anticholinergics (if applied systemically), amantadine, neuroleptics (e. g. phenothiazines) and beta-adrenoceptor agonists (beta-sympathomimetics). Decreased effects due to concomitant medication with cholinergic drugs. Reduced blood pressure in patients treated with isoniazid. The effect of prokinetics such as metoclopramide may be decreased.
Pharmacokinetic interactions are possible with other drugs metabolised by cytochrome P450 3A4 (CYP 3A4). However, a very pronounced increase of concentrations for such drugs is not expected as the effects of propiverine are small compared to classical enzyme inhibitors (e.g. ketoconazole or grapefruit juice). Propiverine may be considered as weak inhibitor of cytochrome P450 3A4. Pharmacokinetic studies with patients concomitantly receiving potent CYP 3A4 inhibitors such as azole antifungals (e.g. ketoconazole, itraconazole) or macrolide antibiotics (e.g. erythromycin, clarithromycin) have not been performed.
4.6 Pregnancy and lactation
There are no adequate data from the use of propiverine hydrochloride in pregnant woman. Studies in animals have shown reproductive toxicity (see section 5.3). The potential risk for humans is unknown.
The drug is secreted into the milk of lactating mammals.
Propiverine hydrochloride should not be used during pregnancy and should not be administered to nursing woman.
4.7 Effects on ability to drive and use machines
No studies on the effects on the ability to drive and use machines have been performed.
Propiverine hydrochloride may produce drowsiness and blurred vision. This may impair the patient’s ability to exert activities that require mental alertness such as operating a motor vehicle or other machinery, or to exert hazardous work while taking this drug.
Sedative drugs may enhance the drowsiness caused by propiverine hydrochloride.
4.8 Undesirable effects
Within each system organ class, the undesirable effects are ranked under heading of frequency using the following convention:
Very common (>1/10)
Common (>1/100 to <1/10)
Uncommon (>1/1,000 to <1/100)
Rare (>1/10,000 to <1/1,000)
Very rare (<1/10,000)
Not known (cannot be estimated from the available data).
Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
Psychiatric disorders
Very rare: restlessness, confusion
Not known: hallucination
Nervous system disorders
Common: headache
Uncommon: tremor, dizziness, dysgeusia
Eye disorders
Common: abnormal accommodation, accommodation disturbances,
abnormal vision
Cardiac disorders
Very rare: palpitation
Vascular disorders
Uncommon: decreased blood pressure with drowsiness, flushing
Gastrointestinal disorders
Very common: dry mouth
Common: constipation, abdominal pain, dyspepsia
Uncommon: nausea/vomiting
Skin and subcutaneous tissue disorders
Rare: rash due to idiosyncrasy (propiverine hydrochloride) or hypersensitivity (excipients)
Renal and urinary disorders
Uncommon: urinary retention
General disorders and administration site conditions
Common:
fatigue
All undesirable effects are transient and recede after a dose reduction or termination of the therapy after maximum 1-4 days.
During long-term therapy hepatic enzymes should be monitored, because reversible changes of liver enzymes might occur in rare cases. Monitoring of intraocular pressure is recommended in patients at risk of developing glaucoma.
Particular attention should be paid to the residual urine volume in cases of urinary tract infections.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via Yellow Card Scheme at: www.mhra.gov.uk/yellowcard
4.9 Overdose
Overdose with the muscarinic receptor antagonist propiverine hydrochloride can potentially result in central anticholinergic effects, e.g. restlessness, dizziness, vertigo, disorders in speech and vision and muscular weakness. Moreover, severe dryness of mucosa, tachycardia and urinary retention may occur.
Treatment should be symptomatic and supportive. Management of overdose may include initiation of vomiting or gastric lavage using an oiled tube (attention: dryness of mucosa!), followed by symptomatic and supportive treatment as for atropine overdose (e.g. physostigmine) with a dosage of 1.0 to 2.0 mg in adults by slow intravenous injection (may be repeated as necessary to a total of 5 mg).
A 14-years old girl who ingested 450 mg propiverine hydrochloride presented with confabulation. The adolescent fully recovered.
5 PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
ATC code: G04BD06
Pharmacotherapeutic group: urinary antispasmodics Mechanism of action
Inhibition of calcium influx and modulation of intracellular calcium in urinary bladder smooth muscle cells causing musculotropic spasmolysis.
Inhibition of the efferent connection of the nervus pelvicus due to anticholinergic action.
Pharmacodynamic effects
In animal models propiverine hydrochloride causes a dose-dependent decrease of the intravesical pressure and an increase in bladder capacity.
The effect is based on the sum of the pharmacological properties of propiverine and three active urinary metabolites as shown in isolated detrusor strips of human and animal origin.
5.2 Pharmacokinetic properties
General characteristics of the active substance
Propiverine is nearly completely absorbed from the gastrointestinal tract. It undergoes extensive first pass metabolism. Effects on urinary bladder smooth muscle cells are due to the parent compound and three active metabolites as well, which are rapidly excreted into the urine.
Absorption
After oral administration of Propinorm® XL 30 mg Capsules propiverine is absorbed from the gastrointestinal tract with maximal plasma concentrations reached after 9.9 hours. The mean absolute bioavailability of Propinorm® XL 30 mg Capsules is 60.8 ± 17.3% (arithmetic mean value ± SD for AUC0^> (p.o.) / AUC0^> (i.v.)).
Food does not influence the pharmacokinetics of propiverine.
The bioavailability of propiverine after the meal was 99% compared to the fasting conditions. Administration of the ER capsule leads to mean Cmax - concentrations of propiverine of about 70 ng/ml reached within 9.5 hours after administration.
The Cmax values for the main metabolite propiverine-N-oxide were slightly increased by food (f= 1.26), whereas the extent of absorption was unchanged. Propiverine-N-oxide showed for all pharmacokinetic parameters 90 % confidence intervals within the acceptance ranges.
An adjustment of dose in relation to food intake is not required.
Distribution
After administration of Propinorm® XL 30 mg Capsules, steady state is reached after four to five days at a higher concentration level than after single dose application(Caverage = 71 ng/ml).
The volume of distribution was estimated in 21 healthy volunteers after intravenous administration of propiverine hydrochloride to range from 125 to 473 l (mean 279 l) indicating, that a large amount of available propiverine is distributed to peripheral compartments. The binding to plasma proteins is 90 - 95 % for the parent compound and about 60 % for the main metabolite.
Pharmacokinetic characteristics (geometric mean, ± SD, range) of propiverine in 10 healthy volunteers after single dose administration of Propinorm® XL 30 mg Modified Release Capsules and propiverine hydrochloride modified release capsules 45 mg:
|
Dose [mg] |
30 |
45 |
|
AUC 0-0O [ng h/ml] |
1378 (903, 2104) |
1909 (1002,3639) |
|
Cmax [ng/ml] |
60.6 (41.5, 88.6) |
80.0 (41.8, 152.1) |
|
t1/2 [h] |
14.2 (10.8, 18.6) |
16.3 (13.9, 19.2) |
|
tmax [h] |
9.9 |
9.9 |
|
± 2.4 |
± 2.4 |
Plasma concentrations of propiverine in 10 healthy volunteers after single dose administration of Propinorm® XL 30 mg Modified Release Capsules and propiverine hydrochloride modified release capsules 45 mg:
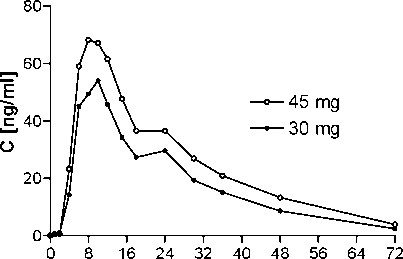
time [h]
Steady state characteristics ofpropiverine following multiple-dose administration to 24 healthy volunteers of propiverine hydrochloride modified release capsules 45 mg once daily for 7 days:
|
geometric mean |
range or ±SD | |||
|
AUC |
0-24h [ngh/ml] |
1711 |
1079, |
2713 |
|
PTF |
[%] |
109.4 |
81.2, |
147.5 |
|
C v-/av |
[ng/ml] |
71 |
45.0, |
113.0 |
|
C '-/max |
[ng/ml] |
105 |
71, |
155 |
|
C v-/mm |
[ng/ml] |
29 |
20, |
42 |
|
t1/2 |
[h] |
20.4 |
12.8, |
32.3 |
|
tmax |
[h] |
7.3 |
± 2.5 | |
PTF: peak-trough fluctuation
Plasma concentrations of propiverine on day 7 and trough levels during treatment following multiple-dose administration of propiverine hydrochloride modified release capsules 45 mg to 24 healthy volunteers once daily for 7 days:
80-
E
O)
c
60-
O O o
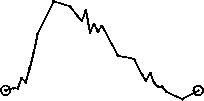
o 40-
20-
o trough levels
0J-1-H h-
-72 -24 0
10
20
30
time [h]
Biotransformation
Propiverine is extensively metabolised by intestinal and hepatic enzymes. The primary metabolic route involves the oxidation of the Piperidyl-N and is mediated by CYP 3A4 and Flavin-monoxygenases (FMO) 1 and 3 and leads to the formation of the much less active N-oxide, the plasma concentration of which greatly exceeds that of the parent substance. Four metabolites were identified in urine; three of them are pharmacologically active and may contribute to the therapeutic efficacy.
In vitro there is a slight inhibition of CYP 3A4 and CYP 2D6 detectable which occurs at concentrations exceeding therapeutic plasma concentrations 10- to 100-fold (see section 4.5).
Elimination
Following administration of 30 mg oral dose of 14C-propiverine hydrochloride to healthy volunteers, 60 % of radioactivity was recovered in urine and 21 % was recovered in faeces within 12 days. Less than 1% of an oral dose is excreted unchanged in the urine. Mean total clearance after single dose administration of 30 mg is 371 ml/min (191 - 870 ml/min).
Linearity/ non-linearity
Pharmacokinetic parameters of propiverine following oral administration of 10 - 45 mg of propiverine hydrochloride are linearly related to dose.
Correlation between the oral dose of extended release propiverine and the resulting AUCQ-cjo:
...O
2000-
q.
•6
r = 0.9961 b = 42.8 a = 27.4
0-f--1-1-1-1-1-1-1-10 10 20 30 40
dose [mg]
Correlation between the oral dose of extended release propiverine and the resulting C ■
max•
o>
= 50-
E
O
25-
..O'
-O'

r = 0.9938 b = 1.72 a = 4.58
0-1-■-1-■-1-■-1-■-r-
0 10 20 30 40
dose [mg]
Characteristics in patients Renal impairment:
Severe renal impairment does not significantly alter the disposition of propiverine and its main metabolite, propiverine-N-oxide, as deduced from a single dose study in 12 patients with creatinine clearance < 30 ml/min. No dose adjustment is to be recommended.
Hepatic insufficiency:
There were similar steady state pharmacokinetics in 12 patients with mild to moderate impairment of liver function due to fatty liver disease as compared to 12 healthy controls. No data are available for severe hepatic impairment.
Age:
The comparison of trough plasma concentrations during steady state reveals no difference between older patients (60 - 85 years; mean 68) and young healthy subjects. The ratio of parent drug to metabolite remains unchanged in older patients indicating the metabolic conversion of propiverine to its main metabolite, propiverine-N-oxide, not to be an age-related or limiting step in the overall excretion. As bioequivalence of Mictonorm® 15 mg Coated Tablets t.i.d. and propiverine hydrochloride modified release capsules 45 mg s.i.d. was established in a GCP compliant study the same can be concluded for Propinorm® XL 30 mg Capsules.
Patients with glaucoma:
The treatment with Propinorm® XL 30 mg Capsules will not lead to an increase of intraocular pressure in patients with open angle glaucoma and in patients with treated (controlled) angle closure glaucoma. This was shown in two placebo-controlled studies with Mictonorm® 15 mg Coated Tablets t.i.d. over 7 days.
5.3 Preclinical safety data
In long term oral dose studies in two mammalian species the main treatment related effect were changes in the liver (including elevation of hepatic enzymes). These were characterised by hepatic hypertrophy and fatty degeneration. The fatty degeneration was reversible upon cessation of treatment.
In animal studies, skeletal retardation in the offspring occurred when the drug was administered orally at high doses to pregnant females. In lactating mammals propiverine hydrochloride was excreted into the milk.
There was no evidence of mutagenicity. The carcinogenicity study in mice demonstrated an increased incidence of hepatocellular adenoma and carcinoma in high dose males. In the rat carcinogenicity study hepatocellular adenoma, kidney
adenoma and urinary bladder papilloma has been demonstrated in high dose male rats, while in female animals endometrial stromal polyps were increased at the high dose levels. Both the rat and mouse tumours were considered to be species specific and therefore not of clinical relevance.
6 PHARMACEUTICAL PARTICULARS
6.1 List of excipients
Pellets Citric acid, povidone,
lactose monohydrate, talc,
triethyl citrate, magnesium stearate,
methacrylic acid-methyl methacrylate copolymer (1:1), methacrylic acid-methyl methacrylate copolymer (1:2), ammonio methacrylate copolymer type A, ammonio methacrylate copolymer type B.
Capsule
Gelatine,
Titanium dioxide E171, red iron oxide E172, yellow iron oxide E172.
6.2 Incompatibilities
Not applicable
6.3 Shelf life
3 years
6.4 Special precautions for storage
Blister:
Store in the original package to protect from moisture. Do not store above 25°C.
Bottle:
Keep the bottle tightly closed.
6.5 Nature and contents of container
Blisters of PVC/PVDC and aluminium foil in cartons of 14, 20, 28, 30, 49, 50, 56, 60, 84, 98, 100, 112 or 280 capsules.
Polyethylene bottles with a polypropylene screw cap containing a silica gel desiccant of 10, 14, 20, 28, 30, 49, 50, 56, 60, 84, 98 or 100 capsules.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal
Not applicable
7 MARKETING AUTHORISATION HOLDER
APOGEPHA Arzneimittel GmbH KyffhauserstraBe 27 01309 Dresden Germany
8 MARKETING AUTHORISATION NUMBER(S)
PL 15072/0007
9 DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
04/05/2006 / 29/06/2011
10 DATE OF REVISION OF THE TEXT
13/06/2014