Spiriva Respimat 2.5 Microgram Inhalation Solution
SUMMARY OF PRODUCT CHARACTERISTICS
1 NAME OF THE MEDICINAL PRODUCT
Spiriva Respimat 2.5 microgram, inhalation solution
2. QUALITATIVE AND QUANTITATIVE COMPOSITION
The delivered dose is 2.5 microgram tiotropium per puff (2 puffs comprise one medicinal dose) and is equivalent to 3.124 microgram tiotropium bromide monohydrate.
The delivered dose is the dose which is available for the patient after passing the mouthpiece.
For the full list of excipients, see section 6.1.
3 PHARMACEUTICAL FORM
Inhalation solution
Clear, colourless, inhalation solution
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
COPD
Tiotropium is indicated as a maintenance bronchodilator treatment to relieve symptoms of patients with chronic obstructive pulmonary disease (COPD).
Asthma
Spiriva Respimat is indicated as an add-on maintenance bronchodilator treatment in adult patients with asthma who are currently treated with the maintenance combination of inhaled corticosteroids (>800 pg budesonide/day or equivalent) and long-acting p2 agonists and who experienced one or more severe exacerbations in the previous year.
4.2 Posology and method of administration
Posology
The medicinal product is intended for inhalation use only. The cartridge can only be inserted and used in the Respimat inhaler (see 4.2).
Two puffs from the Respimat inhaler comprise one medicinal dose.
The recommended dose for adults is 5 microgram tiotropium given as two puffs from the Respimat inhaler once daily, at the same time of the day.
The recommended dose should not be exceeded.
In the treatment of asthma the full benefit will be apparent after several doses of the medicinal product.
Special populations
Geriatric patients can use tiotropium bromide at the recommended dose.
Renally impaired patients can use tiotropium bromide at the recommended dose. For patients with moderate to severe impairment (creatinine clearance < 50 ml/min, see
4.4 and 5.2).
Hepatically impaired patients can use tiotropium bromide at the recommended dose (see 5.2).
Paediatric population COPD
There is no relevant use of Spiriva Respimat in children and adolescents below 18 years
Cystic fibrosis
The efficacy and safety of Spiriva Respimat has not been established (see sections 4.4 and 5.1).
Asthma
The efficacy and safety of Spiriva Respimat in children and adolescents has not yet been established.
Method of administration
To ensure proper administration of the medicinal product, the patient should be shown how to use the inhaler by a physician or other health professionals.
Instructions For Use
Introduction
Spiriva Respimat (tiotropium bromide). Read these Instructions for Use before you start using Spiriva Respimat.
You will need to use this inhaler only ONCE A DAY. Each time you use it take TWO PUFFS.
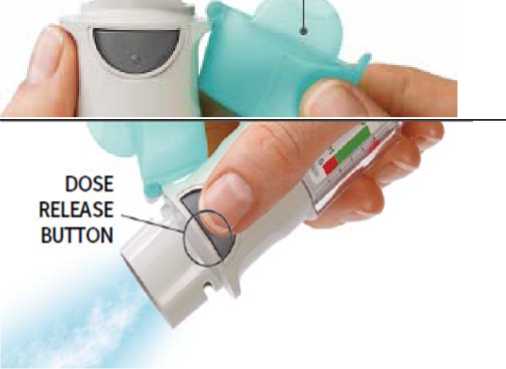
MOUTHPIECE AIR VENT
DOSE-
RELEASE
BUTTON
SAFETY CATCH
CLEAR BASE PIERCING ELEMENT
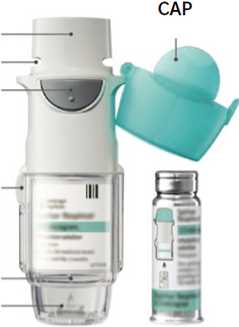
CARTRIDGE
• If Spiriva Respimat has not been used for more than 7 days release one puff towards the ground.
• If Spiriva Respimat has not been used for more than 21 days repeat steps 4 to 6 under ‘Prepare for first use’ until a cloud is visible. Then repeat steps 4 to 6 three more times.
• Do not touch the piercing element inside the clear base.
How to care for your Spiriva Respimat
Clean the mouthpiece including the metal part inside the mouthpiece with a damp cloth or tissue only, at least once a week.
Any minor discoloration in the mouthpiece does not affect your Spiriva Respimat inhaler performance.
If necessary, wipe the outside of your Spiriva Respimat inhaler with a damp cloth.
When to get a new Spiriva Respimat

• Your Spiriva Respimat inhaler contains 60 puffs (30 doses) if used as indicated (two puffs/once daily).
• The dose indicator shows approximately how much medication is left.
• When the dose indicator enters the red area of the scale you need to get a new prescription; there is approximately medication for 7 days left (14 puffs).
• Once the dose indicator reaches the end of the red scale, your Spiriva Respimat locks automatically - no more doses can be released. At this point, the clear base cannot be turned any further.
• Spiriva Respimat should be discarded three months after you have prepared it for first use, even if it has not been fully used or used at all.
Prepare for first use
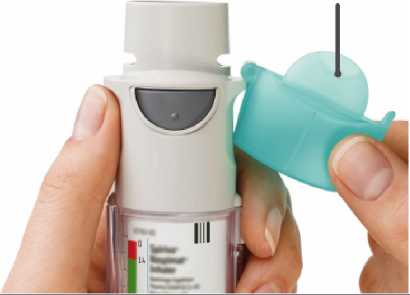
Remove clear base
Keep the cap closed.
Press the safety catch while firmly pulling off the clear base with your other hand.
Insert cartridge
Insert the narrow end of the cartridge into the inhaler. Place the inhaler on a firm surface and push down firmly until it clicks into place.
Do not remove the cartridge once it has been inserted into the inhaler.
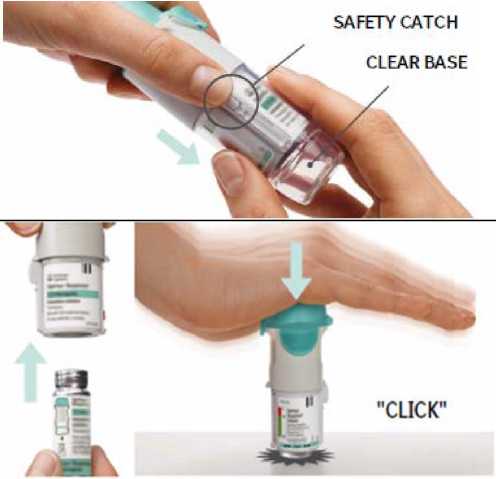
3.
Replace clear base
Put the clear base back into place until it clicks.
Do not remove the clear base again.
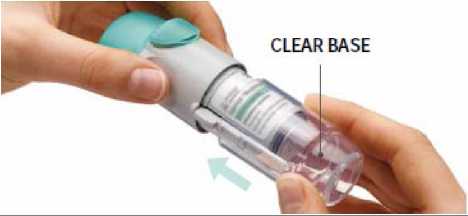
4.
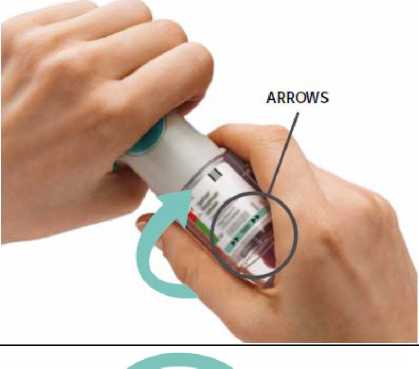
Turn
Keep the cap closed.
Turn the clear base in the direction of the arrows on the label until it clicks (half a turn).
5.
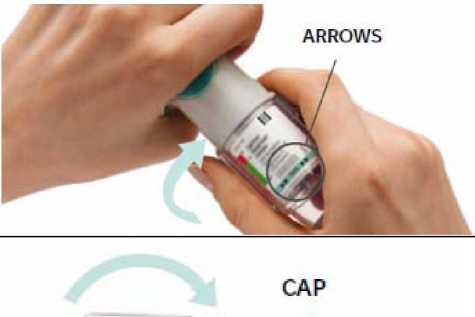
6. Press
• Point the inhaler toward the ground.
• Press the dose-release button.
• Close the cap.
• Repeat steps 4-6 until a cloud is visible.
• After a cloud is visible,
repeat steps 4-6 three more times.
Your inhaler is now ready to use. These steps will not affect the number of doses available. After preparation your inhaler will be able to deliver 60 puffs (30 doses).
TURN
• Keep the cap closed.
• TURN the clear base in the direction of the arrows on the label until it clicks (half a turn).
OPEN
• OPEN the cap until it snaps fully
open.
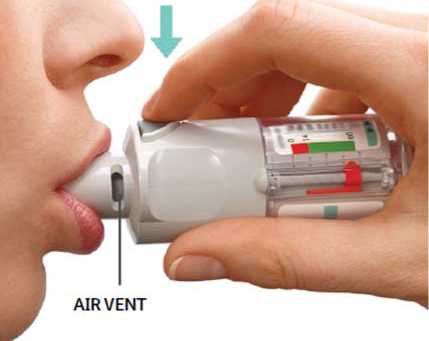
PRESS
• Breathe out slowly and fully.
• Close your lips around the mouthpiece without covering the air vents. Point your Inhaler to the back of your throat.
• While taking a slow, deep breath through your mouth, PRESS the dose-release button and continue to breathe in slowly for as long as comfortable.
• Hold your breath for 10 seconds or for as long as comfortable.
• Repeat Turn, Open, Press for a total of 2 puffs.
• Close the cap until you use your inhaler again.
CAP
Spiriva Respimat is contraindicated in patients with hypersensitivity to tiotropium bromide, atropine or its derivatives, e.g. ipratropium or oxitropium or to any of the excipients (see 6.1).
4.4 Special warnings and precautions for use
Tiotropium bromide, as a once daily maintenance bronchodilator, should not be used for the initial treatment of acute episodes of bronchospasm, or for the relief of acute symptoms. In the event of an acute attack a rapid-acting beta-2-agonist should be used.
Spiriva Respimat should not be used as (first-line) monotherapy for asthma. Asthma patients must be advised to continue taking anti-inflammatory therapy, i.e. inhaled corticosteroids, unchanged after the introduction of Spiriva Respimat, even when their symptoms improve.
Immediate hypersensitivity reactions may occur after administration of tiotropium bromide inhalation solution.
Consistent with its anticholinergic activity, tiotropium bromide should be used with caution in patients with narrow-angle glaucoma, prostatic hyperplasia or bladder-neck obstruction.
Inhaled medicines may cause inhalation-induced bronchospasm.
Tiotropium should be used with caution in patients with recent myocardial infarction < 6 months; any unstable or life threatening cardiac arrhythmia or cardiac arrhythmia requiring intervention or a change in drug therapy in the past year; hospitalisation of heart failure (NYHA Class III or IV) within the past year. These patients were excluded from the clinical trials and these conditions may be affected by the anticholinergic mechanism of action.
As plasma concentration increases with decreased renal function in patients with moderate to severe renal impairment (creatinine clearance < 50 ml/min) tiotropium bromide should be used only if the expected benefit outweighs the potential risk. There is no long term experience in patients with severe renal impairment (see 5.2).
Patients should be cautioned to avoid getting the spray into their eyes. They should be advised that this may result in precipitation or worsening of narrow-angle glaucoma, eye pain or discomfort, temporary blurring of vision, visual halos or coloured images in association with red eyes from conjunctival congestion and corneal oedema.
Should any combination of these eye symptoms develop, patients should stop using tiotropium bromide and consult a specialist immediately.
Dry mouth, which has been observed with anti-cholinergic treatment, may in the long term be associated with dental caries.
Tiotropium bromide should not be used more frequently than once daily (see 4.9).
Spiriva Respimat is not recommended in cystic fibrosis (CF). If used in patients with CF, Spiriva Respimat may increase the signs and symptoms of CF (e.g. serious adverse events, pulmonary exacerbations, respiratory tract infections).
4.5 Interaction with other medicinal products and other forms of interaction
Although no formal drug interaction studies have been performed, tiotropium bromide has been used concomitantly with other drugs commonly used in the treatment of COPD and asthma, including sympathomimetic bronchodilators, methylxanthines, oral and inhaled steroids, antihistamines, mucolytics, leukotriene modifiers, cromones, anti-IgE treatment without clinical evidence of drug interactions.
Use of LABA or ICS was not found to alter the exposure to tiotropium.
The co-administration of tiotropium bromide with other anticholinergic containing drugs has not been studied and therefore is not recommended.
4.6 Fertility, pregnancy and lactation
Pregnancy
There is a very limited amount of data from the use of tiotropium in pregnant women. Animal studies do not indicate direct or indirect harmful effects with respect to reproductive toxicity at clinically relevant doses (see 5.3). As a precautionary measure, it is preferable to avoid the use of Spiriva Respimat during pregnancy.
Breast-feeding
It is unknown whether tiotropium bromide is excreted in human breast milk. Despite studies in rodents which have demonstrated that excretion of tiotropium bromide in breast milk occurs only in small amounts, use of Spiriva Respimat is not recommended during breast-feeding. Tiotropium bromide is a long-acting compound. A decision on whether to continue/discontinue breast-feeding or to continue/discontinue therapy with Spiriva Respimat should be made taking into account the benefit of breast-feeding to the child and the benefit of Spiriva Respimat therapy to the woman.
Fertility
Clinical data on fertility are not available for tiotropium. A non-clinical study performed with tiotropium showed no indication of any adverse effect on fertility (see 5.3).
4.7 Effects on ability to drive and use machines
No studies on the effects on the ability to drive and use machines have been performed. The occurence of dizziness or blurred vision may influence the ability to drive and use machinery.
4.8 Undesirable effects
Summary of the safety profile
Many of the listed undesirable effects can be assigned to the anticholinergic properties of tiotropium bromide.
Tabulated summary of adverse reactions
The frequencies assigned to the undesirable effects listed below are based on crude incidence rates of adverse drug reactions (i.e. events attributed to tiotropium) observed in the tiotropium group pooled from 7 placebo-controlled clinical trials in COPD (3,282 patients) and 6 placebo-controlled clinical trials in asthma (1,256 patients) with treatment periods ranging from four weeks to one year.
Frequency is defined using the following convention:
Very common fej/10); common (>1/100 to <1/10); uncommon fid/1,000 to <1/100); rare (>1/10,000 to <1/1,000); very rare (<1/10,000), not known (cannot be estimated from the available data)
|
System Organ Class / MedDRA Preferred Term |
Frequency COPD |
Frequency Asthma |
|
Metabolism and nutrition disorders | ||
|
Dehydration |
Not known |
Not known |
|
Nervous system disorders | ||
|
Dizziness |
Uncommon |
Uncommon |
|
Headache |
Uncommon |
Uncommon |
|
Insomnia |
Rare |
Uncommon |
|
Eye disorders | ||
|
Glaucoma |
Rare |
Not known |
|
Intraocular pressure increased |
Rare |
Not known |
|
Vision blurred |
Rare |
Not known |
|
System Organ Class / MedDRA Preferred Term |
Frequency COPD |
Frequency Asthma |
|
Cardiac disorders | ||
|
Atrial fibrillation |
Rare |
Not known |
|
Palpitations |
Rare |
Uncommon |
|
Supraventricular tachycardia |
Rare |
Not known |
|
Tachycardia |
Rare |
Not known |
|
Respiratory, thoracic and mediastinal disorders | ||
|
Cough |
Uncommon |
Uncommon |
|
Pharyngitis |
Uncommon |
Uncommon |
|
Dysphonia |
Uncommon |
Uncommon |
|
Epistaxis |
Rare |
Not known |
|
Bronchospasm |
Rare |
Uncommon |
|
Laryngitis |
Rare |
Not known |
|
Sinusitis |
Not known |
Not known |
|
Gastrointestinal disorders | ||
|
Dry Mouth |
Common |
Common |
|
Constipation |
Uncommon |
Rare |
|
Oropharyngeal candidiasis |
Uncommon |
Uncommon |
|
Dysphagia |
Rare |
Not known |
|
Gastrooesophageal reflux disease |
Rare |
Not known |
|
Dental caries |
Rare |
Not known |
|
Gingivitis |
Rare |
Rare |
|
Glossitis |
Rare |
Not known |
|
Stomatitis |
Not known |
Rare |
|
Intestinal obstruction, including ileus paralytic |
Not known |
Not known |
|
Nausea |
Not known |
Not known |
|
Skin and subcutaneous tissue disorders, immune system disorders | ||
|
Rash |
Uncommon |
Rare |
|
Pruritus |
Uncommon |
Rare |
|
Angioneurotic oedema |
Rare |
Rare |
|
System Organ Class / MedDRA Preferred Term |
Frequency COPD |
Frequency Asthma |
|
Urticaria |
Rare |
Rare |
|
Skin infection/skin ulcer |
Rare |
Not known |
|
Dry skin |
Rare |
Not known |
|
Hypersensitivity (including immediate reactions) |
Not known |
Rare |
|
Anaphylactic reaction |
Not known |
Not known |
|
Musculoskeletal and connective tissue disorders | ||
|
Joint swelling |
Not known |
Not known |
|
Renal and urinary disorders | ||
|
Urinary retention |
Uncommon |
Not known |
|
Dysuria |
Uncommon |
Not known |
|
Urinary tract infection |
Rare |
Not known |
Description of selected adverse reactions
In controlled clinical studies in COPD, the commonly observed undesirable effects were anticholinergic undesirable effects such as dry mouth which occurred in approximately 2.9 % of patients. In asthma the incidence of dry mouth was 1.2%.
In 7 clinical trials in COPD, dry mouth led to discontinuation in 3 of 3,282 tiotropium treated patients (0.1 %). No discontinuations due to dry mouth were reported in 6 clinical trials in asthma (1,256 patients).
Serious undesirable effects consistent with anticholinergic effects include glaucoma, constipation, intestinal obstruction including ileus paralytic and urinary retention.
Other special population
An increase in anticholinergic effects may occur with increasing age.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It
allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare
professionals are asked to report any suspected adverse reactions via the Yellow Card Scheme at: www.mhra.gov.uk/yellowcard
Overdose
4.9
High doses of tiotropium bromide may lead to anticholinergic signs and symptoms.
However, there were no systemic anticholinergic adverse effects following a single inhaled dose of up to 340 microgram tiotropium bromide in healthy volunteers. Additionally, no relevant adverse effects, beyond dry mouth/throat and dry nasal mucosa, were observed following 14-day dosing of up to 40 microgram tiotropium inhalation solution in healthy volunteers with the exception of pronounced reduction in salivary flow from day 7 onwards.
5 PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Other drugs for obstructive airway diseases, inhalants, anticholinergics
ATC code: R03B B04
Mechanism of action
Tiotropium bromide is a long-acting, specific antagonist at muscarinic receptors. It has similar affinity to the subtypes, Mj to M5. In the airways, tiotropium bromide competitively and reversibly binds to the M3 receptors in the bronchial smooth musculature, antagonising the cholinergic (bronchoconstrictive) effects of acetylcholine, resulting in bronchial smooth muscle relaxation. The effect was dose dependent and lasted longer than 24h. As an N-quaternary anticholinergic, tiotropium bromide is topically (broncho-) selective when administered by inhalation, demonstrating an acceptable therapeutic range before systemic anticholinergic effects may occur.
Pharmacodynamic effects
The dissociation of tiotropium from especially M3-receptors is very slow, exhibiting a significantly longer dissociation half-life than ipratropium. Dissociation from M2-receptors is faster than from M3, which in functional in vitro studies, elicited (kinetically controlled) receptor subtype selectivity of M3 over M2. The high potency, very slow receptor dissociation and topical inhaled selectivity found its clinical correlate in significant and long-acting bronchodilation in patients with COPD and asthma.
Clinical efficacy and safety in COPD
The clinical Phase III development programme included two 1-year, two 12-weeks and two 4-weeks randomised, double-blind studies in 2901 COPD patients (1038 receiving the 5 pg tiotropium dose). The 1-year programme consisted of two placebo-controlled trials. The two 12-week trials were both active (ipratropium) - and placebo-controlled. All six studies included lung function measurements. In addition, the two 1-year studies included health outcome measures of dyspnoea, health-related quality of life and effect on exacerbations.
Placebo-controlled studies
Lung _ function
Tiotropium inhalation solution, administered once daily, provided significant improvement in lung function (forced expiratory volume in one second and forced vital capacity) within 30 minutes following the first dose, compared to placebo (FEVi mean improvement at 30 minutes: 0.113 litres; 95% confidence interval (CI): 0.102 to 0.125 litres, p< 0.0001). Improvement of lung function was maintained for 24 hours at steady state compared to placebo (FEV1 mean improvement: 0.122 litres; 95% CI: 0.106 to 0.138 litres, p< 0.0001).
Pharmacodynamic steady state was reached within one week.
Spiriva Respimat significantly improved morning and evening PEFR (peak expiratory flow rate) as measured by patient's daily recordings compared to placebo (PEFR mean improvement: mean improvement in the morning 22 L/min; 95% CI: 18 to 55 L/min, p< 0.0001; evening 26 L/min; 95% CI: 23 to 30 L/min, p<0.0001). The use of Spiriva Respimat resulted in a reduction of rescue bronchodilator use compared to placebo (mean reduction in rescue use 0.66 occasions per day, 95% CI: 0.51 to 0.81 occasions per day, p<0.0001).
The bronchodilator effects of Spiriva Respimat were maintained throughout the 1-year period of administration with no evidence of tolerance.
Dyspnoea, Health-related Quality of Life, COPD Exacerbations in long term 1 year studies
Dyspnoea
Spiriva Respimat significantly improved dyspnoea (as evaluated using the Transition Dyspnoea Index) compared to placebo (mean improvement 1.05 units; 95% CI: 0.73 to 1.38 units, p<0.0001). An improvement was maintained throughout the treatment period.
Health-related Quality of Life
The improvement in mean total score of patient’s evaluation of their Quality of Life (as measured using the St. George’s Respiratory Questionnaire) between Spiriva Respimat versus placebo at the end of the two 1-year studies was 3.5 units (95% CI: 2.1 to 4.9, p<0.0001). A 4-unit decrease is considered clinically relevant.
COPD Exacerbations
In three one-year, randomised, double-blind, placebo-controlled clinical trials Spiriva Respimat treatment resulted in a significantly reduced risk of a COPD exacerbation in comparison to placebo. Exacerbations of COPD were defined as “a complex of at least two respiratory events/symptoms with a duration of three days or more requiring a change in treatment (prescription of antibiotics and/or systemic corticosteroids and/or a significant change of the prescribed respiratory medication)”. Spiriva Respimat treatment resulted in a reduced risk of a hospitalisation due to a COPD exacerbation (significant in the appropriately powered large exacerbation trial).
The pooled analysis of two Phase III trials and separate analysis of an additional exacerbation trial is displayed in Table 1. All respiratory medications except anticholinergics and long-acting beta-agonists were allowed as concomitant treatment, i.e. rapidly acting beta-agonists, inhaled corticosteroids and xanthines. Long-acting beta-agonists were allowed in addition in the exacerbation trial.
Table 1: Statistical Analysis of Exacerbations of COPD and Hospitalized COPD Exacerbations in Patients with Moderate to Very Severe COPD
|
Study (NSpiriva, Nplacebo) |
Endpoint |
Spiriva Respimat |
Placebo |
% Risk Reduction (95% CI)a |
p-value |
|
1-year Ph III studies, pooled analysisd (670, 653) |
Days to first COPD exacerbation |
160a |
86a |
29 (16 to 40)b |
<0.0001b |
|
Mean exacerbation incidence rate per patient year |
0.78c |
1.00c |
22 (8 to 33)c |
0.002c | |
|
Time to first hospitalised COPD exacerbation |
25 (-16 to 51)b |
0.20b | |||
|
Mean hospitalised exacerbation incidence rate per patient year |
0.09 c |
0.11 c |
20 (-4 to 38) c |
0.096 c | |
|
1-year Ph IIIb exacerbation study (1939,1953) |
Days to first COPD exacerbation |
169a |
119a |
31 (23 to 37)b |
<0.0001b |
|
Mean exacerbation incidence rate per patient year |
0.69c |
0.87c |
21 (13 to 28)c |
<0.0001c | |
|
Time to first hospitalised COPD exacerbation |
27 (10 to 41)b |
0.003b | |||
|
Mean hospitalised exacerbation incidence rate per patient year |
0.12c |
0.15c |
19 (7 to 30)c |
0.004c |
a Time to first event: days on treatment by when 25% of patients had at least one exacerbation of COPD / hospitalized COPD exacerbation. In study A 25% of placebo
patients had an exacerbation by day 112, whereas for Spiriva Respimat 25% had an exacerbation by day 173 (p=0.09);in study B 25% of placebo patients had an exacerbation by day 74, whereas for Spiriva Respimat 25% had an exacerbation by day 149 (p<0.0001).
b Hazard ratios were estimated from a Cox proportional hazard model. The percentage risk reduction is 100(1 - hazard ratio).
c Poisson regression. Risk reduction is 100(1 - rate ratio).
d Pooling was specified when the studies were designed. The exacerbation endpoints were significantly improved in individual analyses of the two one year studies.
Long-term tiotropium active- controlled study
A long-term large scale randomised, double-blind, active-controlled study with an observation period up to 3 years has been performed to compare the efficacy and safety of Spiriva Respimat and Spiriva HandiHaler (5,711 patients receiving Spiriva Respimat; 5,694 patients receiving Spiriva HandiHaler). The primary endpoints were time to first COPD exacerbation, time to all-cause mortality and in a sub-study (906 patients) trough FEVi (pre-dose).
The time to first COPD exacerbation was numericallysimilar during the study with Spiriva Repimat and Spiriva HandiHaler (hazard ratio (Spiriva Respimat/Spiriva HandiHaler) 0.98 with a 95% CI of 0.93 to 1.03). The median number of days to the first COPD exacerbation was 756 days for Spiriva Respimat and 719 days for Spiriva HandiHaler.
The bronchodilator effect of Spiriva Respimat was sustained over 120 weeks, and was similar to Spiriva HandiHaler. The mean difference in trough FEV1 for Spiriva Respimat versus Spiriva HandiHaler was -0.010 L (95% CI -0.038 to 0.018 L).
In the post-marketing TIOSPIR study comparing Spiriva Respimat and Spiriva HandiHaler, all-cause mortality (including vital status follow up) was similar with hazard ratio (Spiriva Respimat/Spiriva HandiHaler) = 0.96,95% CI 0.84 -1.09). Respective treatment exposure was 13,135 and 13,050 patient-years.
In the placebo-controlled studies with vital status follow-up to the end of the intended treatment period, Spiriva Respimat showed a numerical increase in all-cause mortality compared to placebo (rate ratio (95% confidence interval) of 1.33 (0.93, 1.92) with treatment exposure to Spiriva Respimat of 2,574 patient years; the excess in mortality was observed in patients with known rhythm disorders. Spiriva HandiHaler showed a 13 % reduction in the risk of death ((hazard ratio including vital status follow-up (tiotropium/placebo) = 0.87; 95% CI, 0.76 to 0.99)). Treatment exposure to Spiriva HandiHaler was 10,927 patient-years. No excess mortality risk was observed in the subgroup of patients with known rhythm disorders in the placebo controlled Spiriva HandiHaler study as well as in the TIOSPIR Spiriva Respimat to HandiHaler comparison.
Clinical efficacy and safety in asthma
The clinical Phase III programme for persistent asthma included two 1-year randomised, double-blind, placebo-controlled studies in a total of 907 asthma patients (453 receiving Spiriva Respimat) on a combination of ICS (>800 pg budesonide/day or equivalent) with a LABA. The studies included lung function measurements and severe exacerbations as primary endpoints.
PrimoTinA-asthma studies
In the two 1-year studies in patients who were symptomatic on maintenance treatment of at least ICS (>800 pg budesonide/day or equivalent) plus LABA, Spiriva Respimat showed clinically relevant improvements in lung function over placebo when used as add-on to background treatment.
At week 24, mean improvements in peak and trough FEV1 were 0.110 litres (95% CI: 0.063 to 0.158 litres, p<0.0001) and 0.093 litres (95% CI: 0.050 to 0.137 litres, p<0.0001), respectively. The improvement of lung function compared to placebo was maintained for 24 hours.
In the PrimoTinA-asthma studies, treatment of symptomatic patients (N=453) with ICS plus LABA plus tiotropium reduced the risk of severe asthma exacerbations by 21% as compared to treatment of symptomatic patients (N=454) with ICS plus LABA plus placebo. The risk reduction in the mean number of severe asthma exacerbations/patient year was 20%.
This was supported by a reduction of 31% in risk for asthma worsening and 24% risk reduction in the mean number of asthma worsenings/patient year (see Table 2).
Table 2: Exacerbations in Patients Symptomatic on ICS (>800 pg budesonide/day or equivalent) plus LABA (PrimoTinA-asthma studies)
|
Study |
Endpoint |
Spiriva Respimat, added-on to at least ICSa/LABA (N=453) |
Placebo, added-on to at least ICSa/LABA (N=454) |
% Risk Reduction (95% CI) |
p-value |
|
two 1-year Phase III studies, pooled analysis |
Days to 1st severe asthma exacerbation |
282c |
226c |
(0, 38) |
0.0343 |
|
Mean number of severe asthma exacerbations / patient year |
0.530 |
0.663 |
(0, 36) |
0.0458 | |
|
Days to 1st worsening of asthma |
315c |
181c |
n (18, 42) |
<0.0001 |
|
Study |
Endpoint |
Spiriva Respimat, added-on to at least ICSa/LABA (N=453) |
Placebo, added-on to at least ICSa/LABA (N=454) |
% Risk Reduction (95% CI) |
p-value |
|
Mean number of asthma worsenings / patient year |
2.145 |
2.835 |
""24 (9, 37) |
0.0031 |
a >800 pg budesonide/day or equivalent
b Hazard ratio, confidence interval and p-value obtained from a Cox proportional hazards model with only treatment as effect. The percentage risk reduction is 100(1 -hazard ratio).
c Time to first event: days on treatment by when 25%/50% of patients had at least one severe asthma exacerbation/worsening of asthma
d The rate ratio was obtained from a Poisson regression with log exposure (in years) as offset. The percentage risk reduction is 100 (1-rate ratio).
Paediatric population
COPD
The European Medicines Agency has waived the obligation to submit the results of studies with Spiriva Respimat in all subsets of the paediatric population in COPD (see section 4.2 for information on paediatric use).
Asthma
The European Medicines Agency has deferred the obligation to submit the results of studies with Spiriva Respimat in one or more subsets of the paediatric population in the treatment of asthma (see section 4.2 for information on paediatric use).
Clinical efficacy and safety in cystic fibrosis (CF):
The clinical development programme in CF included 3 multicentre studies in 959 patients aged 5 months and above. Patients below 5 years used a spacer (AeroChamber Plus®) with face mask and were included for safety assessment only. The two pivotal studies (a dose finding Phase II study and a confirmatory Phase III study) compared lung function effects (percent predicted FEVi AUC 0-4h and trough FEVi) of Spiriva Respimat (tiotropium 5 pg: 469 patients) versus placebo (315 patients) in 12-weeks randomised, double-blind periods; the Phase III study also included a long term open label extension, up to 12 months. In these studies, all respiratory medications, except anticholinergics, were allowed as concomitant treatment, e.g. long acting beta agonists, mucolytics and antibiotics.
Effects on lung function are displayed in Table 3. No significant improvement in symptoms and health status (exacerbations by Respiratory and Systemic Symptoms
Questionnaire and quality of life by Cystic Fibrosis Questionnaire) have been observed.
Table 3: Adjusted mean difference from placebo for absolute changes from baseline after 12 weeks
|
Phase II |
Phase III | |||||
|
All patients (Nspiriva = 176, Nplacebo 168) |
All patients (Nspiriva = 293, Nplacebo = 147) |
<11 years (Nspiriva = 95, Nplacebo 47) |
>12 years (Nspiriva = 198, Nplacebo = 100) | |||
|
mean (95% CI) |
p- value |
mean (95% CI) |
p- value |
mean (95% CI) |
mean (95% CI) | |
|
FEV:AUC0-4h (% predicted) a absolute changes |
3.39 (1.67, 5.12) |
<0.00 1 |
1.64 (-0.27, 3.55) |
0.092 |
-0.63 (-4.58, 3.32) |
2.58 (0.50, 4.65) |
|
FEV:AUC0-4h (litres) absolute changes |
0.09 (0.05, 0.14) |
<0.00 1 |
0.07 (0.02, 0.12) |
0.010 |
0.01 (-0.07, 0.08) |
0.10 (0.03, 0.17) |
|
Trough FEVi (% predicted) a absolute changes |
2.22 (0.38, 4.06) |
0.018 |
1.40 -0.50, 3.30 |
0.150 |
-1.24 (-5.20, -271) |
2.56 (0.49, 4.62) |
|
Trough FEV1 (litres) absolute changes |
0.06 (0.01, 0.11) |
0.028 |
0.07 (0.02, 0.12) |
0.012 |
-0.01 (-0.08, 0.06) |
0.10 (0.03, 0.17) |
a Co-primary endpoints
All Adverse Drug Reactions (ADRs) observed in the CF studies are known undesirable effects of tiotropium (see 4.8). The most commonly observed adverse events considered related during the 12 week double blind period were cough (4.1%) and dry mouth (2.8%).
The number and percentage of patients reporting adverse events (AEs) of special interest in cystic fibrosis irrespective of relatedness are shown in Table 4. Signs and symptoms considered to be manifestations of cystic fibrosis increased numerically, although not statistically significantly, with tiotropium, especially in patients <11 years old.
Table 4: Percentage of patients with AEs of special interest in cystic fibrosis by age group
over 12 weeks of treatment irrespective of relatedness (pooled Phase II and Phase III)
<11 years
>12 years
|
Nplacebo 96 |
NSpiriva = 158 |
Nplacebo 215 |
NSpiriva = 307 | |
|
Abdominal pain |
7.3 |
7.0 |
5.1 |
6.2 |
|
Constipation |
1.0 |
1.9 |
2.3 |
2.6 |
|
Distal intestinal obstruction syndrome |
0.0 |
0.0 |
1.4 |
1.3 |
|
Respiratory tract infections |
34.4 |
36.7 |
28.4 |
28.3 |
|
Sputum increased |
1.0 |
5.1 |
5.6 |
6.2 |
|
Exacerbations |
10.4 |
14.6 |
18.6 |
17.9 |
"Distal intestinal obstruction syndrome" and "Sputum increased" are MedDRA preferred terms. "Respiratory tract infections" is the MedDRA higher level group term. "Abdominal pain", "Constipation" and "Exacerbations" are collections of MedDRA preferred terms.
Thirty-four (10.9 %) patients randomised to placebo and 56 (12.0%) patients randomised to Spiriva Respimat experienced a serious adverse event.
The European Medicines Agency has waived the obligation to submit the results of studies with Spiriva Respimat in the subset of paediatric patients below 1 year of age.
5.2 Pharmacokinetic properties
a) General Introduction
Tiotropium bromide is a non-chiral quaternary ammonium compound and is sparingly soluble in water. Tiotropium bromide is available as inhalation solution administered by the Respimat inhaler. Approximately 40% of the inhaled dose is deposited in the lungs, the target organ, the remaining amount being deposited in the gastrointestinal tract. Some of the pharmacokinetic data described below were obtained with higher doses than recommended for therapy.
b) General Characteristics of the Active Substance after Administration of the Medicinal Product
Absorption: Following inhalation by young healthy volunteers, urinary excretion data suggests that approximately 33% of the inhaled dose reaches the systemic circulation. Oral solutions of tiotropium bromide have an absolute bioavailability of 2-3%. Food is not expected to influence the absorption of this quaternary ammonium compound.
Maximum tiotropium plasma concentrations were observed 5-7 minutes after inhalation.
At steady state, peak tiotropium plasma levels in COPD patients of 10.5 pg/ml were achieved and decreased rapidly in a multi-compartmental manner. Steady state trough plasma concentrations were 1.60 pg/ml.
A steady state tiotropium peak plasma concentration of 5.15 pg/ml was attained 5 minutes after the administration of the same dose to patients with asthma.
Systemic exposure to tiotropium following the inhalation of tiotropium via the Respimat inhaler was similar to tiotropium inhaled via the HandiHaler device.
Distribution: The drug has a plasma protein binding of 72% and shows a volume of distribution of 32 l/kg. Local concentrations in the lung are not known, but the mode of administration suggests substantially higher concentrations in the lung. Studies in rats have shown that tiotropium does not penetrate the blood-brain barrier to any relevant extent.
Biotransformation: The extent of biotransformation is small. This is evident from a urinary excretion of 74% of unchanged substance after an intravenous dose to young healthy volunteers. The ester tiotropium bromide is nonenzymatically cleaved to the alcohol (N-methylscopine) and acid compound (dithienylglycolic acid) that are inactive on muscarinic receptors. In-vitro experiments with human liver microsomes and human hepatocytes suggest that some further drug (< 20% of dose after intravenous administration) is metabolised by cytochrome P450 (CYP) dependent oxidation and subsequent glutathion conjugation to a variety of Phase II-metabolites.
In vitro studies in liver microsomes reveal that the enzymatic pathway can be inhibited by the CYP 2D6 (and 3A4) inhibitors, quinidine, ketoconazole and gestodene. Thus CYP 2D6 and 3A4 are involved in metabolic pathway that is responsible for the elimination of a smaller part of the dose.
Tiotropium bromide even in supra-therapeutic concentrations does not inhibit CYP 1A1, 1A2, 2B6, 2C9, 2C19, 2D6, 2E1 or 3A in human liver microsomes.
Elimination: The effective half-life of tiotropium ranges between 27 - 45 h following inhalation by healthy volunteers and COPD patients. The effective half-life was 34 hours in patients with asthma. Total clearance was 880 ml/min after an intravenous dose in young healthy volunteers. Intravenously administered tiotropium is mainly excreted unchanged in urine (74%).
After inhalation of the solution by COPD patients to steady-state, urinary excretion is 18.6 % (0.93 pg) of the dose, the remainder being mainly non-absorbed drug in gut that is eliminated via the faeces. After inhalation of the solution by healthy volunteers urinary excretion is 20.1-29.4 % of the dose, the remainder being mainly nonabsorbed drug in gut that is eliminated via the faeces.
In patients with asthma, 11.9% (0.595 pg) of the dose is excreted unchanged in the urine over 24 hours post dose at steady state. The renal clearance of tiotropium exceeds the creatinine clearance, indicating secretion into the urine.
After chronic once daily inhalation by COPD patients, pharmacokinetic steady-state was reached by day 7 with no accumulation thereafter.
Linearity / Nonlinearity: Tiotropium demonstrates linear pharmacokinetics in the therapeutic range independent of the formulation.
c) Characteristics in Patients
Geriatric Patients: As expected for all predominantly renally excreted drugs, advancing age was associated with a decrease of tiotropium renal clearance (347 ml/min in COPD patients < 65 years to 275 ml/min in COPD patients > 65 years). This did not result in a corresponding increase in AUC0-6ss or Cmax,ss values. Exposure to tiotropium was not found to differ with age in patients with asthma.
Renally Impaired Patients: Following once daily inhaled administrations of tiotropium to steady-state in COPD patients, mild renal impairment (CLcr 50 - 80 ml/min) resulted in slightly higher AUC0-6ss (between 1.8 - 30% higher) and similar Cmax,ss values compared to patients with normal renal function(CLCR >80 ml/min). In COPD patients with moderate to severe renal impairment (CLcr < 50 ml/min), the intravenous administration of a single dose of tiotropium resulted in doubling of the
total exposure (82% higher AUC0-4h) and 52% higher Cmax) compared to COPD patients with normal renal function, which was confirmed by plasma concentrations after dry powder inhalation. In asthma patients with mild renal impairment (CLcr 5080 ml/min) inhaled tiotropium did not result in relevant increases in exposure compared to patients with normal renal function.
Hepatically Impaired Patients: Liver insufficiency is not expected to have any relevant influence on tiotropium pharmacokinetics. Tiotropium is predominantly cleared by renal elimination (74% in young healthy volunteers) and simple nonenzymatic ester cleavage to pharmacologically inactive products.
Japanese COPD Patients: In cross trial comparison, mean peak tiotropium plasma concentrations 10 minutes post-dosing at steady-state were 20% to 70% higher in Japanese compared to Caucasian COPD patients following inhalation of tiotropium but there was no signal for higher mortality or cardiac risk in Japanese patients compared to Caucasian patients. Insufficient pharmacokinetic data is available for other ethnicities or races.
Paediatric Patients:
There were no paediatric patients in the COPD programme (see 4.2). Paediatric patients were studied as part of the CF clinical programme also covering adults.
Following inhalation of 5 pg tiotropium, the tiotropium plasma level in CF patients >5 years was 10.1 pg/ml 5 minutes post-dosing at steady-state and decreased rapidly thereafter. The fraction of the dose available in CF patients <5 years old who used the spacer and mask was approximately 3- to 4-fold lower than that observed in CF patients 5 years and older. Tiotropium exposure was related to body-weight in CF patients <5 years.
d) Pharmacokinetic / Pharmacodynamic Relationship(s)
There is no direct relationship between pharmacokinetics and pharmacodynamics.
5.3 Preclinical safety data
Many effects observed in conventional studies of safety pharmacology, repeat-dose toxicity, and reproductive toxicity could be explained by the anticholinergic properties of tiotropium bromide. Typically in animals reduced food consumption, inhibited body weight gain, dry mouth and nose, reduced lacrimation and salivation, mydriasis and increased heart rate were observed. Other relevant effects noted in repeated dose toxicity studies were: mild irritancy of the respiratory tract in rats and mice evinced by rhinitis and epithelial changes of the nasal cavity and larynx, and prostatitis along with proteinaceous deposits and lithiasis in the bladder in rats.
In juvenile rats exposed from postnatal day 7 to sexual maturity, the same direct and indirect pharmacological changes were observed as in the repeat-dose toxicity studies as well as rhinitis. No systemic toxicity was noted and no toxicologically relevant effects on key developmental parameters, tracheal or key organ development were seen.
Harmful effects with respect to pregnancy, embryonal/foetal development, parturition or postnatal development could only be demonstrated at maternally toxic dose levels. Tiotropium bromide was not teratogenic in rats or rabbits. In a general reproduction and fertility study in rats, there was no indication of any adverse effect on fertility or mating performance of either treated parents or their offspring at any dosage.
The respiratory (irritation) and urogenital (prostatitis) changes and reproductive toxicity was observed at local or systemic exposures more than five-fold the therapeutic exposure. Studies on genotoxicity and carcinogenic potential revealed no special hazard for humans.
6 PHARMACEUTICAL PARTICULARS
6.1 List of excipients
Benzalkonium chloride Disodium edetate Water, purified
Hydrochloric acid 3.6 % (for pH adjustment)
6.2
Incompatibilities
Not applicable.
Shelf life
6.3.
3 years
In-use shelf life: 3 months
6.4 Special precautions for storage
Do not freeze.
6.5 Nature and contents of container
Type and material of the container in contact with the medicinal product:
Solution filled into a polyethylene/polypropylene cartridge with a polypropylene cap with integrated silicone sealing ring. The cartridge is enclosed within an aluminium cylinder.
Pack sizes and devices supplied:
Single pack: 1 Respimat inhaler and 1 cartridge, providing 60 puffs (30 medicinal doses)
Double pack: 2 single packages, each containing 1 Respimat inhaler and 1 cartridge, providing 60 puffs (30 medicinal doses)
Triple pack: 3 single packages, each containing 1 Respimat inhaler and 1 cartridge, providing 60 puffs (30 medicinal doses)
Eight pack: 8 single packages, each containing 1 Respimat inhaler and 1 cartridge, providing 60 puffs (30 medicinal doses)
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
7 MARKETING AUTHORISATION HOLDER
Boehringer Ingelheim International GmbH
Binger Strasse 173 D-55216 Ingelheim am Rhein
Germany
8 MARKETING AUTHORISATION NUMBER(S)
PL 14598/0084
9 DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
24/07/2012
10 DATE OF REVISION OF THE TEXT
11/03/2016