Wilate 500/Wilate 1000 Powder And Solvent For Solution For Injection
SUMMARY OF PRODUCT CHARACTERISTICS
1 NAME OF THE MEDICINAL PRODUCT
Wilate 500, 500 IU VWF/500 IU FVIII, powder and solvent for solution for injection
Wilate 1000, 1000 IU VWF/1000 IU FVIII, powder and solvent for solution for injection
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
Wilate, produced from the plasma of human donors, is presented as a powder and solvent for solution for injection containing nominally 500 IU/1000 IU human von Willebrand factor (VWF) and human coagulation factor VIII (FVIII) per vial.
The product contains approximately 100 IU/ml human von Willebrand factor when reconstituted with 5 ml/10 ml Water for Injections with 0.1 % Polysorbat 80.
The specific activity of Wilate is > 67 IU VWF:RCo/mg protein.
The VWF potency (IU) is measured according to ristocetin cofactor activity (VWF:RCo) compared to the International Standard for von Willebrand Factor Concentrate (WHO).
The product contains approximately 100 IU/ml human coagulation factor VIII when reconstituted with 5 ml/10 ml Water for Injections with 0.1% Polysorbate 80.
The FVIII potency (IU) is determined using the European Pharmacopoeia chromogenic assay. The specific activity of Wilate is > 67 IU FVIIFC/mg protein.
For the full list of excipients, see section 6.1.
3 PHARMACEUTICAL FORM
Powder and solvent for solution for injection.
Freeze-dried powder: white or pale yellow powder or crumbly solid.
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
Von Willebrand disease (VWD)
Prevention and treatment of haemorrhage or surgical bleeding in von Willebrand disease (VWD), when desmopressin (DDAVP) treatment alone is ineffective or contra-indicated.
Haemophilia A
Treatment and prophylaxis of bleeding in patients with haemophilia A (congenital FVIII deficiency).
4.2 Posology and method of administration
Treatment should be under the supervision of a physician experienced in the treatment of coagulation disorders. The product is of single use and the full content of the vial should be administered. In case any content remains, it should be disposed of in accordance with local requirements.
Von Willebrand disease (VWD)
The ratio between VWF:RCo and FVIII:C is 1:1. Generally, 1 IU/kg BW VWF:RCo and FVIII:C raises the plasma level by 1.5-2% of normal activity for the respective protein. Usually, about 20 to 50 IU Wilate/kg BW are necessary to achieve adequate haemostasis. This will raise the VWF:RCo and FVIII:C in the patients by approx. 30 to 100%.
An initial dose of 50 to 80 IU Wilate/kg BW may be required, especially in patients with VWD type 3, where the maintenance of adequate plasma levels may require higher doses than in other types of VWD.
Paediatric population
There are insufficient data to recommend the use of Wilate in children less than 6 years old.
Prevention of haemorrhage in case of surgery or severe trauma:
For prevention of bleeding in case of surgery, Wilate should be given 1-2 hours before start of the surgical procedure. Levels of VWF:RCo of > 60 IU/dl (> 60%) and FVIII:C levels of > 40 IU/dl (> 40%) should be achieved.
An appropriate dose should be re-administered every 12-24 hours of treatment. The dose and duration of the treatment depend on the clinical status of the patient, the type and severity of bleeding, and both VWF:RCo and FVIII:C levels.
In patients receiving FVIII-containing VWF products, plasma levels of FVIII:C should be monitored to reveal sustained excessive FVIII:C plasma levels, which may increase the risk of thrombotic events, particularly in patients with known clinical or laboratory risk factors. In case excessive FVIII:C plasma levels are observed, reduced doses and/or prolongation of the dose interval or the use of VWF product containing a low level of FVIII should be considered.
Prophylaxis:
For long term prophylaxis against bleeds in VWD patients, doses of 20-40 IU/kg bodyweight should be administered 2 or 3 times per week. In some cases, such as in patients with gastrointestinal bleeds, higher doses may be necessary.
Haemophilia A
The dose and duration of the substitution therapy depend on the severity of the FVIII deficiency, on the location and extent of the bleeding and on the patient's clinical condition.
The number of units of FVIII administered is expressed in International Units (IU), which are related to the current WHO standard for FVIII products. FVIII activity in plasma is expressed either as a percentage (relative to normal human plasma) or in IU (relative to an International Standard for FVIII in plasma).
One IU of FVIII activity is equivalent to that quantity of FVIII in one ml of normal human plasma.
On demand treatment:
The calculation of the required dose of FVIII is based on the empirical finding that 1 IU FVIII:C/kg BW raises the plasma level by 1.5-2% of normal activity. The required dose is determined using the following formula:
Required IU = BW (kg) x desired FVIII rise (%) (IU/dl) x 0.5 IU/kg
The amount to be administered and the frequency of administration should always be oriented to the clinical effectiveness in the individual case. In the case of the following haemorrhagic events, the factor VIII activity should not fall below the given plasma activity level (in % of normal or IU/dl) in the corresponding period. The following table can be used to guide dosing in bleeding episodes and surgery.
Treatment scheme for Haemorrhages and Surgery
|
Degree of haemorrhage/ Type of surgical procedure |
FVIII level required (%) (IU/dl) |
Frequency of Doses (hours)/Duration of Therapy (days) |
|
Haemorrhage | ||
|
Early haemarthrosis, muscle bleeding or oral bleeding |
20 - 40 |
Repeat every 12 to 24 hours. At least 1 day, until the bleeding episode as indicated by pain is resolved or healing is achieved. |
|
More extensive haemarthrosis, muscle bleeding or haematoma |
30 - 60 |
Repeat infusion every 12 to 24 hours for 3 to 4 days or more until pain and disability are resolved. |
|
Life threatening haemorrhages |
60 - 100 |
Repeat infusion every 8 to 24 hours until threat is resolved. |
|
Degree of haemorrhage/ Type of surgical procedure |
FVIII level required (%) (IU/dl) |
Frequency of Doses (hours)/Duration of Therapy (days) |
|
Surgery | ||
|
Minor |
30 - 60 |
Every 24 hours, at least 1 |
|
including tooth extraction |
day, until healing is achieved. | |
|
Major |
80 - 100 |
Repeat infusion every 8 to 24 hours until adequate wound healing, then therapy for at |
|
(pre- and postoperative) |
least another 7 days to maintain a FVIII activity of 30% to 60% (IU/dl). |
Prophylaxis:
For long-term prophylaxis against bleedings in patients with severe haemophilia A, doses of 20 to 40 IU Wilate/kg BW should be given at intervals of 2 to 3 days. In some cases, especially in younger patients, shorter dosage intervals or higher doses may be necessary.
Continuous infusion:
Prior to surgery, a pharmacokinetic analysis should be performed to obtain an estimate of clearance. The initial infusion rate can be calculated as follows:
Infusion rate (IU/kg/h) = clearance (mL/kg/h) x desired steady state level (IU/mL)
After the initial 24 hours of continuous infusion, the clearance should be calculated again every day using the steady state equation with the measured level and the known rate of infusion.
During the course of treatment, appropriate determination of FVIII:C levels is advised to guide the dose to be administered and the frequency of repeated infusions. In the case of major surgical interventions in particular, precise monitoring of the substitution therapy by means of coagulation analysis (FVIII:C) is indispensable. Individual patients may vary in their response to FVIII treatment, achieving different half-lives and recoveries.
Previously untreated patients
Currently available data are described in section 4.8.
Paediatric population
There are insufficient data to recommend the use of Wilate in haemophilia A in children less than 6 years old.
Method of administration
Intravenous use.
The injection or infusion rate should not exceed 2-3 ml per minute.
For instructions on reconstitution of the medicinal product before administration, see section 6.6.
4.3 Contraindications
Hypersensitivity to the active substances or to any of the excipients listed in section 6.1.
4.4 Special warnings and precautions for use
As with any intravenous infusion of a plasma-derived protein product, allergic type hypersensitivity reactions are possible. Patients must be closely monitored and carefully observed for any symptoms throughout the infusion period.
Patients should be informed of the early signs of hypersensitivity reactions including hives, generalised urticaria, tightness of the chest, wheezing, hypotension, and anaphylaxis. If allergic symptoms occur, patients should discontinue the administration immediately and contact their physician.
In case of shock, the current medical standards for treatment of shock should be implemented.
Standard measures to prevent infections resulting from the use of medicinal products prepared from human blood or plasma include selection of donors, screening of individual donations and plasma pools for specific markers of infection and the inclusion of effective manufacturing steps for the inactivation/removal of viruses. Despite this, when medicinal products prepared from human blood or plasma are administered, the possibility of transmitting infective agents cannot be totally excluded. This also applies to unknown or emerging viruses and other pathogens.
The measures taken are considered effective for enveloped viruses such as human immunodeficiency virus (HIV), hepatitis B virus (HBV) and hepatitis C virus (HCV), and for the non-enveloped hepatitis A virus. The measures taken may be of limited value against non-enveloped viruses such as parvovirus B19.
Parvovirus B19 infection may be serious for pregnant women (fetal infection) and for individuals with immunodeficiency or increased erythropoiesis (e.g. haemolytic anaemia).
It is strongly recommended that every time that Wilate is administered to a patient, the name and batch number of the product are recorded in order to maintain a link between the patient and the batch of the product.
Appropriate vaccination (hepatitis A and B) should be considered for patients in regular/repeated receipt of human plasma-derived VWF/FVIII concentrates.
Von Willebrand disease (VWD)
When using a FVIII-containing VWF product, the treating physician should be aware that continued treatment may cause an excessive rise in FVIII:C. In patients receiving FVIII-containing VWF products, plasma levels of FVIII:C should be monitored to avoid sustained excessive FVIII:C plasma levels, which may increase the risk of thrombotic events.
There is a risk of occurrence of thrombotic events when using FVIII-containing VWF products, particularly in patients with known clinical or laboratory risk factors. Therefore, patients at risk must be monitored for early signs of thrombosis. Prophylaxis against venous thromboembolism should be instituted, according to the current recommendations.
Patients with VWD, especially type 3 patients, may develop neutralising antibodies (inhibitors) to VWF. If the expected VWF:RCo activity plasma levels are not attained, or if bleeding is not controlled with an appropriate dose, an appropriate assay should be performed to determine if a VWF inhibitor is present. In patients with high levels of inhibitor, VWF therapy may not be effective and other therapeutic options should be considered. Management of such patients should be directed by physicians with experience in the care of patients with haemostatic disorders.
Haemophilia A Hypersensitivity
Allergic type hypersensitivity reactions are possible with Wilate. The product contains traces of human proteins other than FVIII. If symptoms of hypersensitivity occur, patients should be advised to discontinue use of the medicinal product immediately and contact their physician. Patients should be informed of the early signs of hypersensitivity reactions including hives, generalised urticaria, tightness of the chest, wheezing, hypotension, and anaphylaxis.
In case of shock, standard medical treatment for shock should be implemented. Inhibitors
The formation of neutralising antibodies (inhibitors) to FVIII is a known complication in the management of individuals with haemophilia A. These inhibitors are usually IgG immunoglobulins directed against the FVIII procoagulant activity, which are quantified in Bethesda Units (BU) per ml of plasma using the modified assay. The risk of developing inhibitors is correlated to the exposure to FVIII, this risk being highest within the first 20 exposure days. Rarely, inhibitors may develop after the first 100 exposure days.
Cases of recurrent inhibitor (low titre) have been observed after switching from one FVIII product to another in previously treated patients with more than 100 exposure days who have a previous history of inhibitor development. Therefore, it is recommended to monitor all patients carefully for inhibitor occurrence following any product switch.
In general, all patients treated with coagulation FVIII products should be carefully monitored for the development of inhibitors by appropriate clinical observations and laboratory tests. If the expected FVIII activity plasma levels are not attained, or if bleeding is not controlled with an appropriate dose, testing for FVIII inhibitor presence should be performed. In patients with high levels of inhibitor, FVIII therapy may not be effective and other therapeutic options should be considered. Management of such patients should be directed by physicians with experience in the care of haemophilia and FVIII inhibitors.
This medicinal product contains up to 2.55 mmol sodium (58.7 mg) per vial for 500 IU VWF and FVIII/vial and up to 5.1 mmol sodium (117.3 mg) per vial for 1000 IU VWF and FVIII/vial. To be taken into consideration by patients on a controlled sodium diet.
4.5 Interaction with other medicinal products and other forms of interaction
No interactions with other medicinal products are known.
4.6 Fertility, pregnancy and lactation
Animal reproduction studies have not been conducted with VWF/FVIII.
Von Willebrand disease (VWD)
Experience in the treatment of pregnant or lactating women is not available.
Wilate should be administered to pregnant or lactating VWF deficient women only if clearly indicated, taking into consideration that delivery confers an increased risk of haemorrhagic events in these patients.
Haemophilia A
Based on the rare occurrence of haemophilia A in women, experience regarding the treatment during pregnancy and breastfeeding is not available. Therefore, Wilate should be used during pregnancy and lactation only if clearly indicated.
4.7 Effects on ability to drive and use machines
Wilate has no influence on the ability to drive and use machines.
4.8 Undesirable effects
Summary of the safety profile
Hypersensitivity or allergic reactions (which may include angiooedema, burning and stinging at the infusion site, chills, flushing, generalised urticaria, headache, hives, hypotension, lethargy, nausea, restlessness, tachycardia, tightness of the chest, tingling, vomiting, wheezing) have been observed rarely, and may in some cases progress to severe anaphylaxis (including shock). In rare occasions, fever has been observed.
Von Willebrand disease (VWD)
Patients with VWD, especially type 3 patients, may very rarely develop neutralising antibodies to VWF. If such inhibitors occur, the condition will manifest itself as an inadequate clinical response. Such antibodies occur in close association with anaphylactic reactions. Therefore, patients experiencing anaphylactic reaction should be evaluated for the presence of an inhibitor.
In all such cases, it is recommended that a specialised haemophilia centre be contacted.
No cases of inhibitors for von Willebrand factor have been reported from clinical studies or from post marketing experience for Wilate so far.
There is a risk of occurrence of thrombotic events, particularly in patients with known clinical or laboratory risk factors. Prophylaxis against venous thromboembolism should be instituted, according to the current recommendations.
In patients receiving FVIII-containing VWF products sustained excessive FVIII:C plasma levels may increase the risk of thrombotic events.
Haemophilia A
Patients with haemophilia A may develop neutralising antibodies (inhibitors) to FVIII. If such inhibitors occur, the condition will manifest as an insufficient clinical response. In such cases, it is recommended that a specialised haemophilia centre be contacted.
The experience with Wilate in previously untreated patients (PUPs) is limited. In a clinical trial involving 24 PUPs with a minimum of 50 exposure days treated with Wilate, only three patients with a persistent and clinically manifest inhibitor above 5 BU/ml could be detected. Three patients developed low titre, transient inhibitors without any clinical manifestations, and two patients had a low titre inhibitor on a single occasion with no follow-up result.
See also section 4.2. There were no inhibitor developments observed in previously treated patients.
For safety information with respect to transmissible agents, see section 4.4
Tabulated list of adverse reactions
The table presented below is according to the MedDRA system organ classification (SOC and Preferred Term Level).
Frequencies have been evaluated according to the following convention: very common ( 1/10); common (>1/100 to <1/10); uncommon ( 1/1,000 to <1/100); rare ( 1/10,000 to <1/1,000); very rare (<1/10,000), not known (cannot be estimated from the available data).
|
System Organ Class |
Uncommon |
Rare |
Very rare |
|
Immune system disorders |
hypersensitivity reaction |
anaphylactic shock | |
|
General disorders and administration site conditions |
fever | ||
|
Investigations |
FVIII inhibitors |
VWF inhibitors |
Description of selected adverse reactions
For description of selected adverse reactions, see section 4.4
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system. listed in Appendix V - [to be completed nationally].
4.9 Overdose
No symptoms of overdose with human VWF or FVIII have been reported. Thromboembolic events may occur in case of major overdose.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Antihaemorrhagics: blood coagulation factors Von Willebrand factor and coagulation factor VIII in combination ATC Code: B02BD06
Von Willebrand disease (VWD)
The VWF (from the concentrate) is a normal constituent of the human plasma and behaves in the same way as endogenous VWF.
Administration of VWF allows correction of the haemostatic abnormalities exhibited in patients who suffer from VWF deficiency (VWD) at two levels:
- VWF re-establishes platelet adhesion to the vascular sub-endothelium at the site of vascular damage (as it binds both to the vascular sub-endothelium and to the platelet membrane), providing primary haemostasis as shown by the shortening of the bleeding time. This effect occurs immediately and is known to depend to a large extent to the level of polymerisation of the protein;
- VWF produces delayed correction of the associated FVIII deficiency. Administered intravenously, VWF binds endogenous FVIII (which is produced normally by the patient), and by stabilising this factor, avoids its rapid degradation.
Because of this, administration of pure VWF (VWF product with a low FVIII level) restores the FVIII:C level to normal as a secondary effect after first infusion.
Administration of a FVIII-containing VWF preparation restores the FVIII:C level to normal immediately after first infusion.
In addition to its role as a FVIII-protecting protein, VWF mediates platelet adhesion to sites of vascular injury and plays a role in platelet aggregation.
Haemophilia A
The FVIII/VWF complex consists of two molecules (FVIII and VWF) with different physiological functions. When infused into a haemophilia patient, FVIII binds to VWF in the patient's circulation. Activated FVIII (FVIIIa) acts as a cofactor for activated factor IX (FIXa), accelerating the conversion of factor X to activated factor X (FXa). FXa converts prothrombin into thrombin. Thrombin then converts fibrinogen into fibrin and a clot can be formed.
Haemophilia A is a sex-linked hereditary disorder of blood coagulation due to decreased levels of FVIII:C and results in profuse bleeding into joints, muscles or internal organs, either spontaneously or as a result of accidental or surgical trauma. By replacement therapy the plasma levels of FVIII are increased, thereby enabling a temporary correction of the factor deficiency and correction of the bleeding tendencies.
5.2 Pharmacokinetic properties
Von Willebrand disease (VWD)
VWF (from the concentrate) is a normal constituent of the human plasma and acts like the endogenous VWF.
Based on meta-analysis of three pharmacokinetic studies involving 24 evaluable patients with all VWD types, the following results were observed.
|
All VWD types |
VWD type 1 |
VWD type 2 |
VWD type 3 | |||||||||||||||||
|
Parameter |
N |
Mean |
SD |
Min. |
Max. |
N |
Mean |
SD |
Min. |
Max. |
N |
Mean |
SD |
Min. |
Max. |
N |
Mean |
SD |
Min. |
Max. |
|
Recovery (%/IU/kg) |
24 |
1.56 |
0.48 |
0.90 |
2.93 |
2 |
1.19 |
0.07 |
1.14 |
1.24 |
5 |
1.83 |
0.86 |
0.98 |
2.93 |
17 |
1.52 |
0.32 |
0.90 |
2.24 |
|
AUC (0-inf) (h*%) |
23 |
1981 |
960 |
593 |
4831 |
2 |
2062 |
510 |
1701 |
2423 |
5 |
2971 |
1383 |
1511 |
4831 |
16 |
1662 |
622 |
593 |
2606 |
|
T 1/2 (h) |
24 |
23.3 |
12.6 |
7.4 |
58.4 |
2 |
39.7 |
18.3 |
26.7 |
52.7 |
5 |
34.9 |
16 |
17.5 |
58.4 |
17 |
18 |
6.2 |
7.4 |
30.5 |
|
MRT (h) |
24 |
33.1 |
19 |
10.1 |
89.7 |
2 |
53.6 |
25.9 |
35.3 |
71.9 |
5 |
53.5 |
24.6 |
27.8 |
89.7 |
17 |
24.7 |
8.5 |
10.1 |
37.7 |
|
Clearance (mL/h/kg) |
24 |
3.29 |
1.67 |
0.91 |
7.41 |
2 |
2.66 |
0.85 |
2.06 |
3.27 |
5 |
1.95 |
1.02 |
0.91 |
3.31 |
17 |
3.76 |
1.69 |
1.83 |
7.41 |
Key: AUC = area under the curve; MRT = mean residence time
Paediatric population
In a prospective clinical trial involving 15 VWD patients of all types below 6 years of age, recovery assessments were performed in 7 subjects. The median incremental IVR was 0.6 (IQR 1.2) %/IU/kg for VWF:RCo and 1.8 (IQR 1.3) %/IU/kg for FVIII:C.
Haemophilia A
FVIII (from the concentrate) is a normal constituent of the human plasma and acts like the endogenous FVIII. After injection of the product, approximately two thirds to three quarters of the FVIII remain in the circulation. The level of FVIII:C reached in the plasma should be between 80-120% of the predicted FVIII:C.
FVIII:C decreases by a two-phase exponential decay. In the initial phase, distribution between the intravascular and other compartments (body fluids) occurs with a half-life of elimination from the plasma of 3 to 6 hours. In the subsequent slower phase, the half-life varies between 8 to 18 hours, with an average of 15 hours. This corresponds to the true biological half-life.
The following results were observed in one clinical study in 12 patients (chromogenic assay, double measurement):
|
Parameter |
Baseline visit |
6-month visit | ||
|
Mean |
SD |
Mean |
SD | |
|
Recovery %/IU/kg |
FVIII:C 2.27 |
1.20 |
FVIII:C 2.26 |
1.19 |
|
AUCnorm % * h/IU/kg |
FVIII:C 31.3 |
7.31 |
FVIII:C 33.8 |
10.9 |
|
Half-life (h) |
FVIII:C 11.2 |
2.85 |
FVIII:C 11.8 |
3.37 |
|
MRT (h) |
FVIII:C 15.3 |
3.5 |
FVIII:C 16.3 |
4.6 |
|
Clearance mL/h/kg |
FVIII:C 3.37 |
0.86 |
FVIII:C 3.24 |
1.04 |
Key: AUC = area under the curve; MRT = mean residence time; SD = standard deviation
5.3 Preclinical safety data
VWF and FVIII in Wilate are normal constituents of the human plasma and act like the endogenous VWF/FVIII.
Conventional safety testing of these compounds in laboratory animals would not add useful information to the existing clinical experience and therefore is not required.
6 PHARMACEUTICAL PARTICULARS
6.1 List of excipients
Powder: Sodium chloride, Glycine, Sucrose, Sodium citrate and Calcium chloride Solvent: Water for injections with 0.1 % Polysorbate 80
6.2 Incompatibilities
In the absence of compatibility studies, Wilate must not be mixed with other medicinal products or administered simultaneously with other intravenous preparation in the same infusion set.
Only the provided injection/infusion sets should be used because treatment failure can occur as a consequence of FVIII/VWF adsorption to the internal surfaces of some infusion equipment.
6.3 Shelf life
3 years.
The stability of the reconstituted solution has been demonstrated for 12 hours at room temperature (max. +25°C). Nevertheless, to avoid microbial contamination, the reconstituted solution should be used immediately.
6.4 Special precautions for storage
Store powder and solvent vial in a refrigerator (2-8°C). Keep the vials in the outer carton in order to protect from light. Do not freeze.
The product can be stored at room temperature (max. +25°C) for 2 months. In this case the shelf-life expires 2 months after the product has been taken out of the refrigerator for the first time. The new shelf-life has to be noted on the outer carton by the patient. The reconstituted solution should be used on one occasion only. Any solution remaining should be discarded.
For storage conditions after reconstitution of the medicinal product, see section 6.3.
6.5 Nature and contents of container
Package sizes:
Wilate 500, 500 IU VWF and 500 IU FVIII 1 package contains:
1 vial with Powder, type I glass, closed with a stopper (bromobutyl rubber) and sealed with a flip off cap1 vial with Solvent (5 ml Water for Injections with 0.1% Polysorbate 80), type I glass, closed with a stopper (chlorobutyl rubber) and sealed with a flip off cap
1 equipment pack with the medical devices (1 disposable syringe, 1 transfer set [Mix2VialTM], 1 infusion set)
2 alcohol swabs
Wilate 1000, 1000 IU VWF and 1000 IU FVIII 1 package contains:
1 vial with Powder, type I glass, closed with a stopper (bromobutyl rubber) and sealed with a flip off cap
1 vial with Solvent (10 ml Water for Injections with 0.1% Polysorbate 80), type I glass, closed with a stopper (chlorobutyl rubber) and sealed with a flip off cap
1 equipment pack with the medical devices (1 disposable syringe, 1 transfer set [Mix2VialTM], 1 infusion set)
2 alcohol swabs
Not all pack sizes may be marketed.
• Please read all the instructions and follow them carefully!
• Do not use Wilate after expiry date given on the label.
• During the procedure described below, sterility must be maintained!
• The solution in the syringe should be clear or slightly pearly shimmery. Do not inject solutions that are cloudy or have deposits.
• Use the prepared solution immediately, to prevent microbial contamination.
• Only use the injection set provided. The use of other injection/infusion equipment can cause additional risks and treatment failure.
Instructions for Preparing the Solution:
1. Do not use the product directly from the refrigerator. Allow the solvent and the powder in the closed vials to reach room temperature.
2. Remove the flip off caps from both vials and clean the rubber stoppers with one of the provided alcohol swabs.
3. The Mix2vial™ is depicted in Fig. 1. Place the solvent vial on an even surface and hold it firmly. Take the Mix2Vial™ and turn it upside down. Place the blue part of the Mix2Vial™ on top of the solvent vial and press firmly down until it snaps (Fig. 2 + 3).
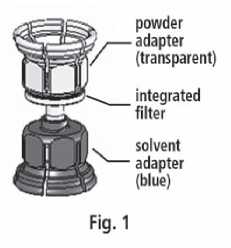
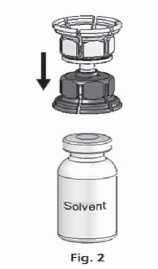
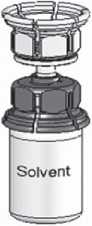
Fig. 3
4. Place the powder vial on an even surface and hold it firmly. Take the solvent vial with the attached Mix2Vial™ and turn it upside down. Place the transparent part on top of the powder vial and press firmly down until it snaps (Fig. 4). The solvent flows automatically into the powder vial.


Fig. 4
5. With both vials still attached, gently swirl the powder vial until the product is dissolved.
The dissolving is completed in less than 10 minutes at room temperature. Slight foaming might occur during preparation. Unscrew the Mix2Vial™ into two parts (Fig. 5). Foaming will disappear.
Dispose the empty solvent vial with the blue part of the Mix2Vial™.

As a precaution, your pulse rate should be taken before and during the injection. If a marked increase in your pulse rate occurs, reduce the injection speed or interrupt the administration for a short time.
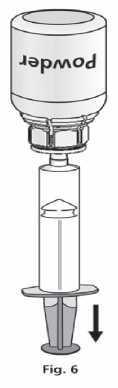
1. Attach the syringe to the transparent part of the Mix2Vial™. Turn the vial upside down and draw the solution into the syringe (Fig. 6).
The solution in the syringe should be clear or slightly pearly shimmery.
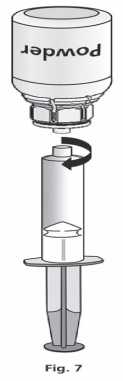
Once the solution has been transferred, firmly hold the plunger of the syringe (keeping it facing down) and remove the syringe from the Mix2Vial™ (Fig. 7). Dispose the Mix2Vial™ and the empty vial.
2. Clean the chosen injection site with one of the provided alcohol swabs
3. Attach the provided injection needle to the syringe.
4. Insert the injection needle into the chosen vein. If you have used a tourniquet to make the vein easier to see, this tourniquet should be released before you start injecting Wilate.
No blood must flow into the syringe due to the risk of formation of fibrin clots.
5. Inject the solution into the vein at a slow speed, not faster than 2-3 ml per minute.
If you use more than one vial of Wilate powder for one treatment, you may use the same injection needle and syringe again. The Mix2Vial™ is for single use only.
Any unused product or waste material should be disposed of in accordance with local requirements.
7 MARKETING AUTHORISATION HOLDER
To be completed nationally
8 MARKETING AUTHORISATION NUMBER(S)
To be completed nationally
9 DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
To be completed nationally
10 DATE OF REVISION OF THE TEXT
Last revision: 12.05.2014